
Симптоми, ознаки і лікування синдрому Щасливої ляльки (Ангельмана)
Хвороба Ангельмана (синдром петрушки) – Орфа розлад генетичної і неврологічної природи.
Ця патологія вперше була виділена і описана в профільній літературі англійським лікарем Гаррі Ангельманом в 1965 році, що і зумовило одна з назв захворювання.
Іншими позначеннями хвороби є такі терміни як «синдром Петрушки» або «синдром щасливою ляльки».
Чому ляльки і що це означає? Зазначені назви зумовлені специфічною моторикою, особливостями поведінки і виразу обличчя (фото дітей див. Нижче), які характерні для людей з хворобою Ангельмана. Нижче – розглянемо їх детально.
Синдром петрушки що це
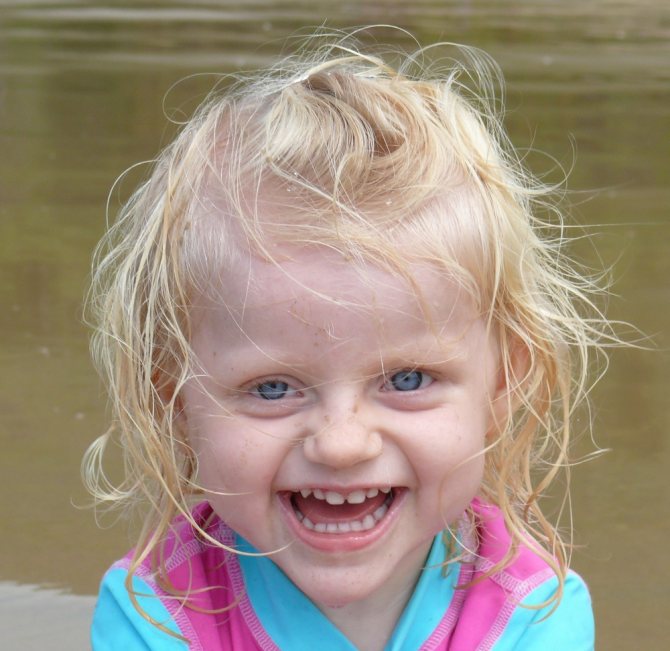
Найбільш поширений вік діагностики захворювання становить від трьох до семи років, коли ознаки хвороби Ангельмана стають найбільш очевидними. Немовлята здаються нормальними при народженні, але часто мають проблеми з годуванням в перші місяці життя, виявляють помітні затримки розвитку на 6-12 місяців.
Напади часто починаються від 2 до 3 років. Порушення мови виражено, часто проявляється гіперактивність, малий розмір голови, розлади сну і моторики (атаксія), неспровокована епізодами сміху і посмішки.
Генетичні мутації підвищують ризик появи захворювання
Поява синдрому Ангельмана пов’язано з наявністю у батьків майбутньої дитини різних хромосомних аномалій. Серед таких відхилень зазвичай називають:
- трисомії хромосом – присутність однієї або декількох зайвих хромосом в хромосомному наборі;
- інверсію – розворот одного з ділянок хромосоми на 180 градусів, при цьому частина хромосоми пропущена, а гени розташовуються в протилежному порядку;
- мікроделеції, яка є результатом перебудови Y-хромосоми і обміну ділянками між хромосомами, спостерігається невелика кількість хромосом, а також може бути відсутнім один з генів;
- делецию – брак одного з ділянок хромосоми;
- транслокацию – перенесення або приєднання ділянки однієї хромосоми до іншої хромосоми;
- дуплікацію – копіювання частини хромосом, результатом чого стає зайвий генетичний матеріал;
- кільцеву хромосому – на кінцях хромосоми відсутня генетичний матеріал, при цьому новостворені кінці з’єднуються в вигляді кільця.
Генні мутації, які можуть викликати розвиток синдрому
Симптоми і ознаки
Розлад пов’язано з широким спектром можливих симптомів. Конкретні симптоми синдрому Ангельмана варіюються від випадку до випадку. Особи з патологією не матимуть всіх симптомів, про які йде мова нижче. Наприклад, у деяких людей можуть бути судоми, у інших – ні. Більшість нездатні говорити, а іноді є обмежена мова.
Діти з синдромом Ангельмана відчувають затримки в досягненні етапів розвитку (затримки розвитку) і мають серйозні труднощі при навчанні. Більшість не розвивають здатності говорити більше, ніж кілька слів. Зазвичай розуміють прості команди.
Старші діти і дорослі можуть спілкуватися за допомогою жестів і / або використання комунікаційних карт, але є порушення висловлювання думок.
ранні ознаки
При ранньому виявленні помітні порушення руху або балансу, уривчасті руху через нездатність їх координувати (атаксія). Має місце зменшений м’язовий тонус (гіпотонія) тулуба, збільшений м’язовий тонус (гіпертонус) рук, ніг і аномально перебільшені або жваві рефлекторні реакції (гіперрефлексія).
Порушення руху проявляються під час дитинства (приблизно 6-12 місяців). Моторні етапи (наприклад, ходьба) зазвичай затримуються. У легких випадках діти починають ходити у віці 2-3 років.
При важких випадках, ходьба може бути помітно повільної, жорсткою і уривчасто. Деякі не можуть ходити, аж поки не збудеться 5-10 років. Приблизно в 10 відсотках випадків діти з синдромом Ангельмана не ходять без сторонньої допомоги.
У новонароджених
У новонародженого і дітей з хворобою Ангельмана є виразні симптоми, характерного щасливого поведінки з частими, недоречними епізодами неспровоцированного, тривалого сміху. Діти можуть бути легко порушено, гіпермоторние і гіперактивні. Вони є активними дослідниками, постійно в русі.
У багатьох випадках виникають епілептичні припадки. Напади зазвичай починаються від одного до п’яти років і часто поліпшуються в підлітковому віці.
Деякі прояви, пов’язані з синдромом сміється ляльки, зустрічаються рідше, ніж вищезгадані симптоми.
Риси обличчя
У деяких випадках індивідууми мають характерні риси обличчя, видатний підборіддя, глибокі очі, аномально широкий рот (маркостомія) з виступаючим мовою, широко розставленими зубами і аномально плоскою задньою частиною голови (брахицефалия).
Шлунково-кишкові
Проблеми з харчуванням і сном виникають в дитинстві через проблеми з ковтанням, порушень руху мови. Діти або дорослі часто відчувають запор або гастроезофагеальний рефлюкс-розлад (ГЕРБ), стан, що характеризується зворотним потоком вмісту шлунка або тонкого кишечника в трубку, яка з’єднує рот зі шлунком (стравохід).
додаткові симптоми
Надмірне слинотеча, схрещені очі (косоокість), відсутність нормального кольору (гипопигментации) шкіри, очей і волосся через відсутність певних меланінових пігментів.
Це відсутність пігменту в очах може викликати чутливість до світла (світлобоязнь), швидке, мимовільне рух очей (ністагм) і зниження ясності зору (гострота зору).
Порушення сну, такі як зниження потреби у сні і порушення або ненормальні цикли сну / неспання (наприклад, пробудження в нічний час або раніше, ніж зазвичай) є частими явищами. Діти люблять воду, музику, їх приваблюють блискучі предмети.
У деяких – підвищена чутливість до тепла. З віком може стати очевидною прогресивна кривизна спинного хребта (сколіоз). Статеве дозрівання зазвичай не затримується, дітонародження можливо.
Дорослі мають більш виражені риси обличчя, нижньощелепний прогнатизм. Іноді розвивається аномальне випинання рогівки (кератоконус). Рухливість зменшується у міру дорослішання, розвиваються спазми суглобів (контрактури). Деякі схильні до ожиріння.
Симптоми, як правило присутні
- Нормальна пренатальна і родинна історія, нормальна окружність голови при народженні, відсутність серйозних вроджених дефектів.
- Нормальні метаболічні, гематологічні та хімічні лабораторні профілі.
- Структурно нормальний мозок на магнітно-резонансної томографії (МРТ) або комп’ютерної томографії. Може спостерігатися легка атрофія кори або дісміелінізація.
- Відстрочене досягнення етапів розвитку без втрати навичок.
- Затримки розвитку від 6 до 12 місяців, класифікуються як тяжкі.
- Порушення мови, з мінімальним використанням слів; навички невербальної комунікації вище, ніж виразні мовні навички.
- Порушення руху або балансу, зазвичай атаксія ходи і / або тремтяче рух кінцівок.
- Поведінкова унікальність, включаючи будь-яку комбінацію частого сміху / посмішки; збудливість, часто з ручними рухами; гіпермоторние поведінку; дефіцит уваги.
Більш ніж у 80% постраждалих
- Відстрочений або непропорційно повільне зростання окружності голови. Зазвичай призводить до абсолютної або відносної микроцефалии до віку 2 років.
- Судоми, зазвичай починаючи з 3 років.
- Аномальна електроенцефалограма (ЕЕГ) з характерною структурою повільних хвиль з великою амплітудою.
Менш ніж у 80%
- Плоский потилицю.
- Потиличний паз.
- Виступаючий мову.
- Смоктальні / ковтальні розлади.
- Проблеми з годуванням і / або м’язова гіпотонія в дитинстві.
- Prognathia.
- Широкий рот, широко розставлені зуби.
- Часте слинотеча.
- Надмірне поведінку при жуванні / блювота.
- Косоокість.
- Гіпопігментовані шкіра, світле волосся та колір очей (у порівнянні з сім’єю); видно тільки в тих, у кого є видалення.
- Гіперактивні рефлекси глибоких сухожиль нижніх кінцівок.
- Підняте, зігнуте положення руки, особливо під час руху.
- Широка хода з пронацією або вальгус щиколотки.
- Підвищена чутливість до тепла.
- Аномальні цикли сну і неспання, зниження потреби у сні.
- Захоплення водою; чарівність хрусткими предметами, такими як папір, пластмаси.
- Аномальна поведінка, пов’язане з харчуванням.
- Ожиріння (частіше зустрічається у тих, у кого немає виключення).
- Сколіоз.
- Запор.
Етіологія захворювання
Даний синдром зустрічається досить рідко, приблизно в 1 випадку на 10-20 тисяч новонароджених. Основною причиною захворювання є втрата копії нормальних генів матері в 15 хромосомі, що відбувається внаслідок порушення процесу ділення цієї хромосоми. Крім цього, захворювання може виникнути внаслідок мутації генів по лінії батька, батьківської трисомії або дісоміі.
У нормі у здорової людини спостерігається передача по одній копії 15 хромосома від матері і від батька. Якщо ж дитина отримає від одного з батьків кілька генетично змінену копію (особливо якщо це ген матері, оскільки вони сильніші в порівнянні з генами батька), то у малюка буде розвиватися синдром Ангельмана.
причини
Дефіцит експресії гена E3 ubiquitin protein ligase (UBE3A) викликає синдром Ангельмана. Ген – частина 15 хромосоми (15q11-q13).
Хромосоми, які присутні в ядрі клітин людини, несуть генетичну інформацію. Клітини людського тіла зазвичай мають 46 хромосом.
Пари хромосом людини пронумеровані від 1 до 22, а статеві хромосоми позначені X і Y. У хлопчиків одна Х і одна Y-хромосома, у дівчаток дві Х-хромосоми.
Кожна хромосома має коротку руку, позначену «р», а довгу руку позначають «q». Хромосоми далі поділяються на багато груп, які нумеруються.
Наприклад, «хромосома 15q11-q13» відноситься до смуг 11-13 на довгому плечі хромосоми 15. Пронумеровані смуги визначають місце розташування тисяч генів, присутніх на кожній хромосомі.
мутації
Мутації в гені ube3a, що викликають синдром Англмана, пов’язані з відсутністю гена, змінами в структурі гена або змінами функції, експресії гена. Приблизно в 10 відсотках випадків ніяка причина не може бути ідентифікована.
У більшості випадків генетичні зміни є результатом спонтанної мутації, але приблизно в 3-5% їх можна успадкувати
.
У приблизно 70-75 відсотках випадків відбувається мікроделеції області 15q11-13 материнської хромосоми 15, яка включає делецию гена UBE3A.
Дізнатися більше Всі симптоми синдрому Сотоса
Це видалення зазвичай відбувається спорадично (de novo) і не успадковується.
Ризик народження дитини з синдромом Ангельмана в сім’ї оцінюється 1-2 відсотка або менше.
Унікальним генетичним феноменом, пов’язаним з синдромом маріонетки, є «імпринтинг». Кожен має дві копії кожного гена: один отриманий від батька, а інший від матері. У більшості випадків обидва гена включаються і активні.
Однак в деяких випадках один ген переважно блокується або відключається в залежності від того, від якого батька він з’явився. Цей процес інактивації «батьківського походження» є прикладом «геномного імпринтингу».
Особливості
Приблизно в 20 відсотках випадків (3-5%) це викликано видаленням ДНК в Центрі импринтинга; інші 80 відсотків випадків викликані поки невідомими або непізнаними дефектами генетичного імпринтингу.
Може бути до 50 відсотків ризику повторення синдрому Ангельмана через импринтинга дефектів, які мають делеції ДНК.
Деякі люди з симптомами синдрому Ангельмана не мають ідентифікованої аномалії хромосоми 15. Деякі індивідууми в цій групі можуть мати розлад, відмінне від синдрому петрушки.
Простими словами
Нижче наведено дуже загальне пояснення, написане для батьків.
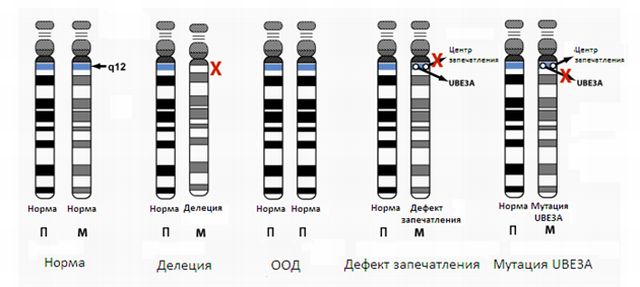
- Ілюстрація хромосоми 15 з червоним розділом, який містить ген UBE3a, і часто видаляється з материнської хромосоми при синдромі ляльки.
- Існує багато механізмів, які викликають розлад. Найбільш поширеним є «видалення».
Якщо 15-я хромосома схожа на те в енциклопедії, зі спеціальною главою, яка знаходиться тільки в обсязі, успадкованому від матері, тоді уявіть, що ця глава була вирвана.
Більшість людей з синдромом сміється ляльки мають видалення в хромосомі 15. У їхньому обсязі відсутня відсутня глава, тому мозку не вистачає деяких інструкцій, які потрібно розвивати.
від батька
Глава батька присутній, але «вимита» через природного процесу імпринтингу, тому сторінки виглядають порожніми і клітини мозку не можуть отримати доступ до інформації.
- Інші люди мають мутацію в гені UBE3A. Це схоже на неправильне написання в важливою чолі, яка присутня тільки на хромосомі, успадкованої від матері. Це неправильне правопис настільки серйозне, що робить велику частину голови нерозбірливою для клітин у мозку. Знову ж, кінцевим результатом є те, що мозку не вистачає важливої інформації для навчання та розвитку.
- У деяких людей з синдромом Петрушки є одностороння діссоматія або UPD. У цих випадках людина успадкував дві копії цього «томи 15» від батька і без копії від матері. Знову ж таки, інформація, яка знаходиться тільки в обсязі, успадкованому від матері, відсутня, і обидві копії від батька порожні або «вимиті».
- У невеликого числа людей з Angelman є дефект відбитка центру (IC) на материнській хромосомі. На 15-й хромосомі існує область, яка допомагає хромосомі вирішити, чи доступні глави інформації або «вимиті».
від матері
Коли є дефект в материнській ІС, глави, які зазвичай доступні, стають недоступними. Незважаючи на те, що «обсяг 15» є повним і нормальним, клітини мозку не можуть отримати доступ до необхідної їм інформації.
- На цьому малюнку показані різні механізми, які викликають розлад. Червоні хромосоми представляють собою хромосому, успадковану від матері, а сині – батьком.
У першому прикладі показана нормальна пара «обсягу» 15 з хромосомної главою батька з «розмитою» або «мовчить» UBE3A, в той час як глава матері активна, а UBE3A робить генетичні інструкції в цій області доступними клітинам головного мозку.
інші приклади
Інші приклади ілюструють відомі механізми, які викликають патологію, роблячи генетичну інформацію матері недоступною для клітин в головному мозку.
Наприклад, при делециях ген UBE3A повністю відсутня, оскільки «глава» була «вирвана». У мутаціях UBE3A ген UBE3A «неправильно прийнятий», що робить цю копію UBE3A нефункціональної.
- Нарешті, у кількох людей є всі або більшість симптомів синдрому Ангельмана, але кожен тест на їх гени виявляється нормальним. У багатьох з цих випадків цілком ймовірно, що у нас просто немає технології і досвіду.
Все, що ми знаємо напевно, це те, що UBE3A має життєво важливе значення для розвитку мозку, контролю мови, руху і навчання. Дослідження майбутніх методів лікування Angelman спирається на пошук способу зробити UBE3A присутнім в батьківському «обсязі» доступним для мозку.
По суті, якщо ми думаємо про те, що генетична інформація про хромосомі батька «розмита» і невидима, вчені шукають способи, щоб дані знову з’являлися, мозок міг отримати доступ до інформації, а люди з AS могли нормально розвиватися.
Видалення 15q11.2-q13 (~ 68% випадків)
Більшість випадків AS викликані делеціями на материнській копії хромосоми 15. Таким чином, делеция усуває нормальну експресію цього гена у індивідуумів AS.
Мутації UBE3A (~ 11% випадків)
У цих людей мутації в гені UBE3A або запобігають його експресію, яку функцію. Таким чином, ці люди не мають відповідних рівнів функціонального UBE3A в головному мозку.
Uniparental disomy (UPD, ~ 7% випадків)
У UPD у індивіда є дві копії батьківської хромосоми 15. Оскільки UBE3A не виражає з батьківської копії, у цих людей немає нормального рівня UBE3A в мозку.
Відбиток дефекту (~ 3% випадків)
Ці індивідууми можуть мати видалення імпрінтінгового центру на хромосомі 15, але випадки можуть також бути викликані втратою імпрінтінговой інформації під час оогенезу матері. Втрата импринтинга запобіжить експресію материнського гена UBE3A в головному мозку.
Клінічні / інші (~ 11%)
У цих людей все тести нормальні, але вони як і раніше відповідають діагностичним критеріям для AS. Ці індивідууми можуть мати досі не визнані мутації, які впливають на UBE3A або геномної імпрінтінг на хромосомі 15.
Зверніть увагу, що існує кілька інших синдромів, які представляють собою AS, які можуть бути протестовані.
Синдром Ангельмана – каріотип
Діагноз ставиться способом генетичного аналізу. Каріотип при синдромі Ангельмана схематично виглядає як 46 XX або XY, 15q-. Раптовий хромосомний збій є відсутність значної суміжній галузі з великого комплексу (до 4 млн) пар основ ДНК в ділянці q11-q13 ділянок 15-ї хромосоми. Деякі альтернативні дослідники визначають причиною розвитку дефекту вторинну мутацію гена UBE3A. Ферментна компонента комплексної системи деградації білків (убіквітин) – продукт цієї мутації.
діагностика
Діагноз – синдром Ангельмана робиться на основі докладної історії хвороби, ретельної клінічної оцінки та ідентифікації характерних симптомів.
Близько 80% випадків виявляються за допомогою різних спеціалізованих аналізів крові, таких як метилювання ДНК (виявляє, але не робить відмінностей між видаленням хромосом, дефектом відбитка і уніпаратарной дезоманіей).
Флуоресцентна гібридизація in situ (FISH) або порівняльна геномна гібридизація виявляють характерну делецию (спостережувану в 70% випадків) однієї копії 15 хромосоми в клітинах організму.
Мутаційний аналіз гена Angelman, UBE3A, виявляє близько 10% випадків, що мають негативні дослідження метилування ДНК.
тестування
Типи тестування доступні для різних причин синдрому щасливою ляльки.
- Тест на метилювання ДНК визначить ~ 80% людей з синдромом Ангельмана, в тому числі з делеціями, UPD і порушеннями імпринтингу. Якщо людина старше і відповідає критеріям, спочатку може бути замовлено FISH-тестування, після цього необхідно провести тестування метилування, щоб визначити, чи є проблема материнської або батьківської.
- Якщо аналіз FISH і метилування виглядає нормальним, рекомендується секвенувати ген UBE3A безпосередньо для визначення потенційних мутацій.
- Зверніть увагу, що 11% дітей, які відповідають діагностичним критеріям будуть давати нормальні показники у всіх доступних тестах. Ці індивідууми можуть мати невизнані мутації, які впливають на UBE3A або геномної імпрінтінг на хромосомі 15.
Зверніть увагу, що існує кілька схожих синдромів.
Диференціальний діагноз
Симптоми наступних розладів можуть бути подібними. Порівняння корисні для диференціальної діагностики.
Немовлята з синдромом Ангельмана зазвичай демонструють неспецифічну психомоторну затримку або судоми, тому диференційний діагноз часто широкий і неспецифічний.
Паралельно розглядають церебральний параліч, статичну енцефалопатію або мітохондріальну енцефаломіопатія, аутистические розлади. Трикутність і різкі рухи кінцівок, які спостерігаються у більшості дітей з синдромом Петрушки, допомагають його відрізнити від цих станів.
Список розладів, які можна прийняти за синдром Ангельмана
Prader-Willi Syndrome – є результатом делеції на хромосомі 15, яка успадкована по батьковій лінії. Оскільки цей представляє серйозну гіпотонію і труднощі з годуванням, він може бути неправильно діагностовано як синдром Ангельмана і навпаки.
Дізнатися більше Способи лікування і ознаки хвороби Шарко марі тута, невральної аміотрофії
Х-пов’язаний синдром розумової відсталості (XLMR), тип Крістіансон. Цей синдром викликаний мутаціями в гені SLC9A6 і згадується як «подібний англійцям». До клінічних особливостей відносяться, щаслива аффективность, серйозні когнітивні затримки, атаксія, мікроцефалія, судорожне розлад. У деяких буває атрофія мозочка і головного мозку.
Люди з розладом SLC9A6 часто мають більш худий зовнішній вигляд і втрачають амбулаторно після 10 років.
Синдром Ретта – розлад нервової системи, яке відбувається майже виключно у дівчаток. Характеризується розумовою відсталістю, судомами, втратою мови і регресією придбаних навичок.
Хромосома 22q13.3 Phelan-McDermid (синдром Фелан-Макдермід). Стан проявляється недіссоморфнимі рисами обличчя, відсутністю або мінімальною промовою, помірної або важкої затримкою розвитку, іноді з поведінковими особливостями в спектрі аутизму, гіпотонією.
Моват-Вілсон – складне розлад розвитку, що характеризується розумовою відсталістю, затримкою розвитку, мови, моторики, епілепсію, щасливим афекту, мікроцефалією і запором. Зазвичай є результатом гетерозиготних мутацій в ZEB2.
Моносомія 1p36 – видалення області 1p36 призводить до множинних вроджених аномалій, розумової відсталості.
Синдром Сміта-Магеніса – видалення областей 17p11.2 призводить до розумової відсталості, гіпотонії, затримки мови, порушень сну.
Chromosome 17q21.31 Microdeletion Syndrome – призводить до розумової відсталості, гіпотонії і доброзичливому розташуванню з характерними лицьовими аномаліями.
Синдром Пітта-Хопкінса
Викликаний гаплоінтенсівностью гена TCF4 на 18q21. Він проявляється поведінковими характеристиками. Наприклад, відсутність мовлення, мікроцефалія, судоми, атаксична хода і щасливі посмішки, але також має відмінні риси.
Це розумова інвалідність, широкий рот, відмінні риси обличчя, також переривчаста гіпервентиляція з наступним апное. Добова гіпервентиляція є характерною особливістю, виникає після трирічного віку.
Chromosome 2q23.1 Синдром мікроделеції – він був недавно описаний в Pubmed; він включає мікроцефалія, судоми і короткий зростання. У доповіді говориться, що люди, які спочатку вважалися синдромом Ангела, Прадера-Віллі або Сміт-Магенісом, позитивно показали це видалення.
Інші порушення мікроделеції, особливо нові, виявлені порівняльної геномної гибридизацией (аналіз хромосомних мікрочіпів), можуть бути пов’язані з деякими особливостями синдрому щасливою ляльки.
мутація HERC2
Відома як MRT38 – аутосомно-рецесивна, розумова відсталість. Порушення функції HERC2 пов’язано зі зменшенням активності E6-AP, що призводить до затримки в глобальному розвитку та аутістіческім функцій, подібним з AS. Особи можуть також мати сині райдужки (очі).
Дефіцит аденілосукціната ліази призводить до накопичення сукцінілпурінов, що призводять до психомоторної затримки, аутістіческім особливостям, гіпотонії, судом. Повідомлялося про моторну апраксии, важкому мовному дефіциті, надмірному сміху, дуже щасливому положенні, гіперактивності, дефіциті уваги, ковтанні предметів, істериці і стереотипних рухах.
Особливості виховання і адаптації
У міру дорослішання дитини з даною патологією симптоми змінюються, стають менш інтенсивними, або навпаки більш інтенсивними, можуть зникати старі і з’являтися нові. Перспективи в стані хворого багато в чому залежать від умов, які оточують хворого.
Доброзичлива среда, атмосфера любові ласки дає шанс хворому стати, принаймні, частково самостійною людиною. При належному догляді ознаки хвороби з часом стають легше. Тривалість життя у людей з синдромом Ангельмана середня. Необхідно пам’ятати про регулярні обстеження, правильне харчування, а також дотримання умов терапії.
Навчання дитини в школі вимагає застосування інклюзивних програм. Однак в України і країнах СНД на сьогоднішній день ці програми не входять в широку практику. Таким дітям необхідний особливий підхід, що включає в себе навчання концентрації уваги. Важливою вимогою також є можливість навчального закладу надати медичну допомогу при нападі.
У нашому суспільстві на даний момент до людей з хромосомними порушеннями ставляться з обережністю і побоюванням. В останні роки активно проводяться різні заходи, спрямовані на поширення знань про таких захворюваннях, а також на формування толерантності в суспільстві.
лікування
Терапія синдрому Ангельмана є симптоматичної та підтримуючої. Кілька клінічних випробувань тривають, але поки немає ніякої генетичної терапії або лікувальних препаратів. Загальна фізичне здоров’я дітей, що мають синдром щасливою ляльки – добрий. Показана звичайна педіатрична допомога, звичайна імунізація.
протисудомну
Пацієнти з синдромом Ангельмана, які відчувають напади, потребують препаратів проти судом (протисудомних). Зазвичай судоми адекватно контролюються за допомогою одних ліків. При деяких випадках контроль утруднений, і необхідно кілька ліків.
Було доведено, що жоден протисудомний препарат не є найбільш ефективним у всіх випадках. Порушення сну є загальними і потребують дотримання строгих ритуалів сну, поведінкової терапії. Іноді потрібно застосування заспокійливих ліків.
Проблеми годування і шлунково-кишкового тракту
Проблеми годування новонароджених вирішуються за допомогою модифікованих методів грудного вигодовування або таких засобів, як спеціальні соски, щоб допомогти дітям з поганою здатністю смоктати.
Гастроезофагеальний рефлюкс лікується за допомогою вертикального позиціонування і ліків, що допомагають переміщенню їжі по травній системі (препарати рухливості).
У деяких випадках може знадобитися хірургічне затягування клапана, який з’єднує стравохід зі шлунком (сфінктер стравоходу). Для лікування запору використовуються проносні засоби.
Опорно-рухові
Дужки / опори щиколотки і фізіотерапія допомагають навчитися ходити. Сколіоз розвивається приблизно в 10% випадків, тоді потрібні дужки або хірургічна корекція. Часто косоокість коригується хірургічно.
Раннє сприяння важливо для забезпечення того, щоб діти з синдромом Ангельмана досягли свого потенціалу. Спеціальні послуги включають спеціальну підтримку, медичну і психологічну допомогу. Більшість дітей отримують фізичну, мовну та психологічну терапію. Корекційна поведінкова терапія застосовується для запобігання небажаної поведінки.
Генетична консультація принесе користь сім’ям з синдромом Ангельмана.
Підводячи підсумок:
Спеціальної терапії немає. Зазвичай необхідна медикаментозна допомога при нападах. Фізична, комунікаційна, поведінкова терапії дозволяють людям досягти максимального потенціалу розвитку.
Фото і відео матеріали по темі
Відео, а також галерея з фото дітей і дорослих, у яких діагностовано Синдром Ангельмана:

Дитина з синдромом ляльки Дорослий з синдромом Ангельмана
Прогноз і профілактика
Більшість людей, що мають синдром щасливою маріонетки (Ангельмана), матимуть серйозні затримки розвитку, обмеження мови і рухові труднощі.
Проте, вони можуть мати нормальне життя, і не регресувати в розвитку в міру старіння. Рання діагностика та індивідуальні методи лікування допомагають поліпшити якість життя.
Вони, потребують довічне догляді, інтенсивної терапії, ретельному медичному нагляді.
Важливо відзначити, що втрата UBE3A, не впливає на розвиток нейронів, що вказує на те, що вони можуть нормально функціонувати, якщо функція UBE3A відновлюється.
прогноз
Ступінь хромосомних порушень безпосередньо впливає на характер розвитку хвороби і на розвиток пацієнта. Іноді діти опановують навички спілкування і самостійної ходьби, в такому випадку можна сказати про те, що їх стан не важкий. В інших випадках дієздатність відсутня протягом усього життя.
Купірування симптомів в більшості випадків з часом дає свій ефект, в зрілому віці пацієнти сплять спокійніше, гіперактивність йде на другий план, знижується частота аномалій ЕЕГ, нападів судом, іноді вони повністю зникають. Протилежної ситуація може бути серед пацієнтів жіночої статі, у яких з настанням статевого дозрівання збільшується частота нападів.
В цілому можна говорити про хороший стан здоров’я і середньої тривалості життя від двадцяти до п’ятдесяти років. Якість же останньої безпосередньо залежить від тяжкості патології. Підвищена увага і догляд з боку близьких залишиться пріоритетною умовою успішного лікування.
Додатковими позитивними факторами стане дотримання призначених лікарем умов терапії, правильного раціону харчування і проходження регулярних обстежень. Належний догляд вбереже від ускладнень. Максимального комфорту для пацієнта можна досягти за умови відповідального поводжень батьків.
Яким буде дитина з синдромом ляльки?
Можливо, ви помітили, що ваш малюк має затримку в розвитку і звернулися за допомогою до педіатра. Через те, що це рідкісне захворювання, лікарі зазвичай не підозрюють синдром Ангельмана у дітей.
Коли батькам говорять, діагноз – синдром сміється ляльки, вони не знайомі з характеристиками цього розладу. Наприклад, судоми, проблеми зі сном, відхилення в поведінці, сміх, невербальне спілкування, важкі когнітивні порушення, затримка в розвитку мови.
Ранній діагноз корисний. По-перше, у вас більше часу, щоб дізнатися про те, що значить виховувати дитину з особливими потребами. Це може бути страшним, оскільки є багато чинників, які слід враховувати, це: спеціальну освіту; лікарі та вузькі фахівці.
Діагностика також означає ранній початок терапії та доступ до послуг, які, мають величезну користь для малюка.
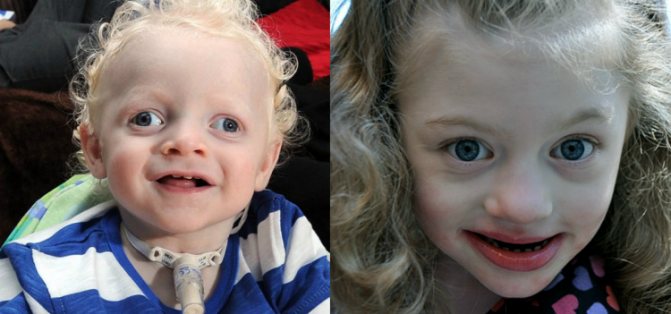
Не менш важлива і пізня діагностика. Хоча батьки / опікуни можуть підозрювати, що їх дитина має особливі потреби, отримання генетичного діагнозу відкриває двері для широкої бази знань і стратегій, які є ефективними в АС, а також для доступу до товариства людей мають подібний досвід.
часті питання
Як буде виглядати дитина? Це природне запитання – чи зможе він сидіти або ходити. Ви можете запитати: «Чи буде дитина говорити або використовувати мову жестів? Як буде грати з іншими дітьми? Чи піде в школу? Чи зможе їздити на велосипеді? Як буде розвиватися? »
Хоча ніхто не може передбачити це точно, ми знаємо, що не всі діти з AS відчувають однаковий ступінь і тяжкість симптомів. Ваша дитина є одночасно продуктом ~ 29 000 нормально функціонуючих генів.
Незважаючи на відсутність «лікування» генетичного дефекту, фізична, психологічна, мовна терапія корисні для дітей з затримкою розвитку незалежно від генотипу або актуального розвитку
Помилково припускати, що всі характеристики, будуть присутні у кожної людини до ступеня, описаної клініцистами для синдрому в цілому.
Оцінюючи досягнення в області розвитку або прогрес в таких областях, як «коли дитина почала сидіти, ходити, грати в ігри». Різниця велика навіть серед дітей з аналогічним генотипом.
особливості харчування
Людині з синдромом Ангельмана потрібно поживна дієта. З огляду на проблеми з годуванням і рефлюксом, корисно звернутися за порадою до дієтологів. Якщо дитина не може приймати тверду їжу, батьки повинні проконсультуватися з медичним працівником про G-подібній трубці, що забезпечує поживні речовини через хірургічне отвір в шлунку.
Заключна думка, полягає в тому, що, хоча людина народжується з втратою функції одного гена, UBE3A (ген «Angelman»), існують тисячі інших повністю функціональних. Так, діагноз стане зміною життя, але багато інших чинників впливають на розвиток і визначають її загальну якість.
Найкраще, що може зробити будь-який батько, – це отримати інформацію. Знайдіть відмінного лікаря. Шукайте інших батьків і приєднуйтесь до організації синдрому Ангельмана, яка найкращим чином відповідає вашим цінностям.
Дрібні чудеса відбуваються кожен день. Залишайте двері відкритими, щоб впустити їх.
(Всі люди, зображені в цій статті, мають синдром Ангельмана).
Поліпшення стану і життя хворих
Синдром Ангельмана є генетичним захворюванням, на сьогоднішній день не існує методів відновлення хромосомних порушень, і лікування неможливо. Однак існує велика кількість способів зменшення симптоматики, що полегшує стан таких людей.
У кожному конкретному випадку програма реабілітації розробляється індивідуально, відповідно симптомів і станом конкретного пацієнта. Фахівці виділяють чотири основні напрями терапії:
- Прийом протиепілептичних препаратів і антиконвульсантів. Дані препарати допомагають контролювати і знижувати частоту нападів, викликаних захворюванням.
- Лікувальна фізкультура – допомагає розвивати дрібну моторику і вирішувати інші проблеми рухового апарату. Діти з аномаліями хромосом розвиваються повільніше своїх здорових однолітків, і це вимагає терпіння з боку сім’ї.
- Мова жестів. Хворі з синдромом маріонетки мало розмовляють, проте цілком успішно використовують мову жестів. Навчання варто почати з самого раннього віку.
- Поведінкова терапія. Дана програма дозволяє дати коректне і ефективне виховання дітей, які мають відхилення, допоможе впоратися з гіперактивністю і дефіцитом уваги.
Багато лікарів відзначають схожість між дітьми, що страждають на аутизм і синдром Ангельмана. Американськими вченими були досягнуті успіхи в лікуванні, а саме застосування внутрішньовенних ін’єкцій з гормоном Секретин. Позитивний ефект виражається в зменшенні ознак небажаної поведінки, а також поліпшенні комунікативних навичок.
ускладнення
Синдром Ангельмана – рідкісне маловідоме захворювання. У зв’язку з цим постановка діагнозу і надання медико-психолого-педагогічної допомоги часто проводяться несвоєчасно, до 6-8 років, що обумовлює низьку успішність корекційних та лікувальних заходів. Без фізіотерапевтичного лікування поглиблюються порушення опорно-рухового апарату – хворі страждають від важких форм сколіозу, самостійно пересуваються з працею. Своєрідність зовнішності і поведінки стає причиною погіршення і без того утрудненою соціальної адаптації. Все перераховане тягне за собою ускладнення інвалідизації пацієнтів.
Як лікується «синдром Ельфа»
Хворі з діагнозом «синдром Вільямса (особа Ельфа)» лікування отримують лише у вигляді симптоматичної терапії, так як генетичні захворювання в наш час не мають специфічного лікування.
Для малюків, які потрапили до лікарні з гіперкальціємією у важкій формі, призначають глюкокортикоїди ( «Гідрокортизон»), а дорослим хворим зазвичай рекомендують постійне спостереження за рівнем кальцію в крові і споживанням вітаміну D, під впливом якого рівень кальцію збільшується.
Дітям з названим синдромом потрібні посилені заняття з психологом. Контакт зі здоровими дітьми також має для таких пацієнтів велике значення, так як створює для них позитивну динаміку в загальному розвитку.
На що слід звертати увагу
Батьки в першу чергу, а також лікар, повинні звертати увагу на наступні моменти в поведінці дитини:
- Рвучкі і хаотичні рухи.
- Дуже часто дитина сміється без причин.
- Неприродне вираз обличчя.
- Тремтіння рук і ніг.
- При ходьбі ноги практично не згинаються в колінах.
- Дуже погано переносять спеку.
- Язик висунутий назовні.

Якщо дитина має дивний вираз обличчя, часто язик висунутий з рота, то варто показати його фахівцеві
Як розпізнати синдром Вільямса ( «особа Ельфа»)
Крім характерного зовнішнього вигляду, у хворих синдромом Вільямса відзначені і особливості розвитку вищих нервових функцій, які призводять до виникнення специфічних особливостей в психіці.
- Порушення сенсорної інтеграції (процесу, при якому нервова система отримує сигнали від дотику, вестибулярного апарату, нюху, зору, смаку і слуху). При цьому чутливість до звуку у пацієнтів підвищена.
- Хворі імпульсивні, легко збудливі, дуже товариські (іноді навіть нав’язливо), настрій у них нестійка (в медицині дана особливість характеризується як емоційна лабільність).
- У них спостерігається підвищена тривожність, помітний страх всього нового.
- Характерні порушення здатності вимови звуків (експресивна мова) і сприйняття сказаного (импрессивная мова).
- Під час навчання таких хворих помітні проблеми з засвоюванням математичних дій.
При цьому те, як проявляється затримка розумового розвитку у таких дітей, виглядає незвично. Їх мова, як правило, добре розвинена для їх віку, виразна і забарвлена емоційно, крім того, у них спостерігаються явні музичні здібності: абсолютний слух, відмінне почуття ритму і прекрасна музична пам’ять. Вони можуть довго старанно займатися грою на музичних інструментах або прослуховуванням творів, але з працею зосереджуються на будь-яких інших речах.

форми
Синдром Ангельмана пов’язаний з чотирма варіантами генетичних мутацій:
- Знову виникла хромосомної мутацією, яка пов’язана з втратою ділянки хромосоми в локусі 15 q11 – q13. Дана мутація – причина близько 80% всіх випадків захворювання.
- Одноотцовской дісоміі, яка пов’язана з втратою материнського локусу (відсутність генетичного матеріалу матері). Даний варіант зустрічається рідко (близько 5% всіх випадків).
- Дефектом ряду генів, схильних до геномному імпринтингу (ГІ). Дані дефекти виникають у 2-4% хворих в результаті безпосереднього порушення імпринтингу (відмінності в перетворенні інформації гена в білок або РНК, які залежать від походження гена). Найчастіше виникає в результаті втрати центру регуляції ГІ. Дефекти ГІ без втрати центру регуляції є результатом спонтанної мутації, повторення якої – велика рідкість.
- Спонтанної мутацією материнської копії, яка викликає відсутність перетворення в мозку копії гена UBE3A. Даний ген кодує діяльність убіквітінлігази (фермент, який бере участь в складному процесі розпаду білків). Дефіцит даного ферменту відноситься до молекулярних механізмів синдрому.
Встановити форму захворювання у 7-9% в даний час не представляється можливим.
Клінічна характеристика дитини
Зовні хвора дитина виглядає цілком задоволеним і щасливим, однак, це враження оманливе.
У таких дітей завжди відзначається сильна затримка розумових і фізичних показників, що накладає свій негативний відбиток на навчання і спілкування з однолітками.
У важких випадках розвивається стійке і значне порушення моторики рук, що робить дитину нездатним самостійно про себе піклуватися (зокрема, дитина не може самостійно застебнути ґудзик або блискавку на одязі або взуття, вчиняти будь-які інші дії, які здоровій людині здаються цілком буденними) .
Все це істотно погіршує якість життя дитини.
У дітей, які страждають синдромом Ангельмана, відзначаються і зовнішні відмінності, такі як:
- пласке обличчя (середня його частина), недостатній розвиток лицьових кісток черепа;
- маленький і загострене підборіддя;
- гіпертрофована нижня щелепа, яка сильно видається вперед;
- великий і широкий мову, який дитина часто висовує назовні;
- порушення зубного ряду, наявність міжзубних проміжків;
- маленький череп (інші кістки скелета мають нормальні розміри, відповідні віковим показниками);
- деформація (викривлення) хребта.
Фізичні характеристики Синдрому Ангелмана
Так само, як синдром Дауна можна розпізнати за специфічними відхиленнями голови і обличчя, синдром Ангельмана характеризується непропорційно малим колом голови. Симптом зазвичай не спостерігається під час народження, а тільки в міру розвитку дитини, протягом якого голова не може рости разом з іншим тілом.
Це призводить до мікроцефалії, станом, при якому мозок ненормально малий. На відміну від деяких форм микроцефалии, які проявляються при народженні (наприклад, новонароджені, врожденно інфіковані вірусом Зика), ті, які викликані синдромом Ангельмана, розпізнаються тільки в віці від одного до двох років.
На додаток до розміру голови, інші характерні симптоми можуть включати в себе:
- Брахіцефалія (плоский потилицю)
- Telecanthus (широко розставлені очі)
- Двосторонні епікантіческіе складки (помітні шкірні складки на верхніх і нижніх повіках)
- Косоокість (косоокість)
- Макростомія (широкий рот)
- Широко розставлені зуби
- Конічні пальці з широкими великими пальцями
- Гладкі долоні з ненормальними складками
- Гіпопігментація шкіри, волосся або очей (відсутність кольору)
Синдром Ангелмана не пов’язаний ні з аномальним зростанням, ні розміром кінцівок, ні з статевим розвитком. Статева зрілість і фертильність як у чоловіків, так і у жінок не будуть зачіпатися. Менструація і сперматогенез (розвиток сперматозоїдів в період статевого дозрівання) відбуваються більш-менш в той же час, що і у інших дітей.
У міру дорослішання дітей з синдромом Ангельмана може спостерігатися прогресуюча латеральна кривизна хребта (сколіоз). У деяких дорослих також розвивається макрогнатії (аномальне розширення щелепи) і кератоконус (випинання рогівки).
Ожиріння також поширене, особливо у дорослих жінок.
Ще кілька цікавих особливостей «Ельфів»
Дослідники, які вивчають синдром Вільямса ( «особа Ельфа»), причини, симптоми цього захворювання, виявили, що страждають їм люди мають особливу вибірковістю в сприйнятті візуально отриманої інформації.

Так, якщо показати даному пацієнтові фото велосипеда і запропонувати перемалювати зображення, то він зробить це з великою точністю, не упускаючи жодної дрібниці. Правда, кермо, колеса і педалі будуть розкидані по всьому листу. Зате в разі, коли це ж завдання потрібно виконати із зображенням обличчя людини, то він виявиться на висоті, примудрившись, крім того, по фотографії не гірше хорошого психолога вловити характер людини.