
Синдром Шерешевського-Тернера – причини, симптоми, діагностика та способи лікування
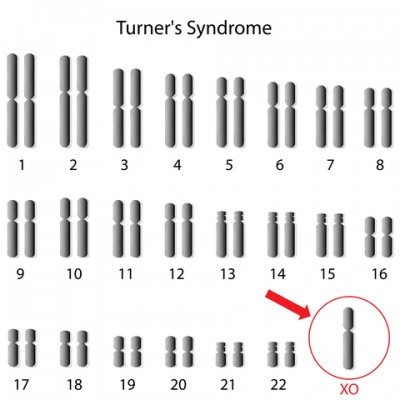
Синдром Шерешевського-Тернера – генетично детермінована аномалія, що характеризується порушенням психофізичного статусу, недорозвиненням геніталій і низьким зростанням. Цей вроджений недуга розвивається у дівчаток, що мають одну статеву X-хромосому замість двох. Характерний каріотип хворої людини – 45Х0, але зустрічаються й інші варіанти. Дитина зазвичай народжується недоношеною з цілим рядом небезпечних відхилень. Часткова або повна Х-моносомія накладає відбиток на все подальше життя хворих дітей.
Синдром проявляється недорозвиненням вторинних статевих ознак, молочних залоз, гипогонадизмом, наявністю аномалій внутрішніх органів. У пацієнток часто повністю відсутні яєчники і місячні, волосся росте на грудях і особі, виявляються вроджені патології нирок, серця і судин, суглобові контрактури, шкірні складки на короткій шиї, набряклі кінцівки. Хворі відстають від своїх однолітків в моторному і статевому розвитку. Зовнішній вигляд дорослих жінок відрізняється грубими рисами обличчя, ростом волосся по шиї до самої спини, широким розрізом очей, деформацією вух, Х-образними ногами, низьким ростом.
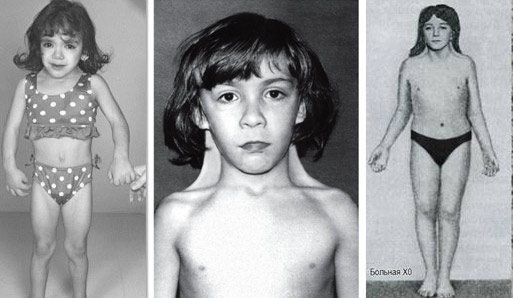
З раннього віку хворі діти відчувають, що вони не схожі на інших і усвідомлюють свою неповноцінність. У дівчаток виробляється комплекс через маленькі груди, недоліків фігури і низькорослості. Але деяка інфантильність, що зберігається навіть в зрілому віці, допомагає хворим легше сприймати свої вади і переносити постійне психоемоційне перенапруження. Правильний підхід до дітей з синдромом Тернера допомагає їм швидше адаптуватися в суспільстві, вести повноцінне життя, дружити, вчитися, закохуватися, заводити сім’ї. Інтелектуальний розвиток у більшості з них не має відхилень.
Діагностика синдрому грунтується на характерних клінічних даних та результатах цитогенетичного аналізу. В даний час проводять пренатальну діагностику недуги за допомогою УЗД плоду і інвазивних методик. Хворих лікують гормональними препаратами, проводять хірургічне відновлює і загальнозміцнюючий лікування.
Синдром був відкритий на початку минулого століття ендокринологом Н. А. Шерешевський, який описав симптоми вродженого гіпогонадизму у своїх пацієнток. Через кілька років фахівець в галузі ендокринології Тернер інакше визначив характер патології і довів, що статевий інфантилізм завжди поєднується із зовнішніми проявами і суглобовими деформаціями. Відомо, що на 3000 новонароджених народжується 1 хвора дитина. Але ці статистичні дані є досить умовними, оскільки на ранніх термінах у вагітних часто трапляються викидні. Синдром має код по МКБ-10 Q96 і найменування «Синдром Тернера».
Що це таке?
Аномалія діагностується переважно у дівчаток, у осіб чоловічої статі присутній лише при мозаїчному типі патології.
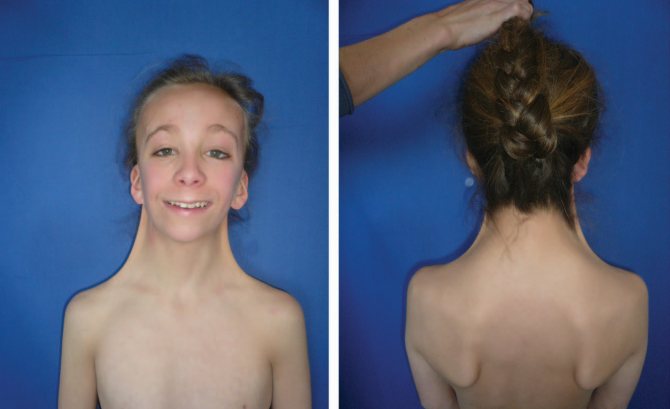
Хвороба виникає у представників всіх без винятку рас.
Для синдрому Ульріха-Тернера характерно відсутність в каріотипі новонароджених однієї з статевих хромосом.
Першим патологію описав в 1925 році лікар-ендокринолог Н. Шерешевський. У 1938 році доктор Г. Тернер виділив основні ознаки недуги. У 1959 році Ч. Форд дізнався природу порушення.
діагностичні заходи
Діагностикою та лікуванням синдрому займаються генетики, гінекологи, неонатологи, педіатри, а також лікарі вузьких спеціальностей. Перш за все звертають на себе увагу характерні клінічні ознаки, наявні навіть у новонароджених дітей. Синдром зі стертою клінічною картиною виявляється в пубертатному періоді по відсутності менархе, недорозвинення статевих органів.
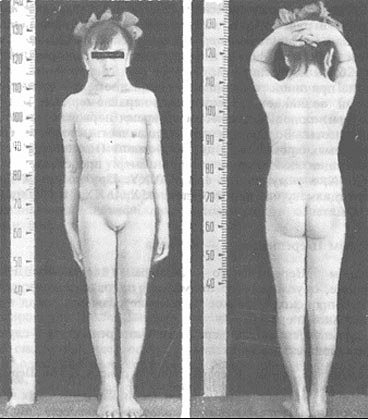
Лабораторні та інструментальні методики:
- Гормональне дослідження крові – підвищення гонадотропінів і зниження естрогенів.
- Молекулярно-генетичне дослідження – вивчення каріотипу і визначення статевого хроматину.
- Цитогенетичний аналіз і дослідження з Y-специфічним зондом – верифікація відсутності Х-хромосоми або її структурних змін.
- Пренатальна діагностика полягає у виявленні ознак синдрому у плоду за даними УЗД або інвазивних методів: біопсії хоріона, амниоцентеза.
- ЕхоКГ і ЕКГ виявляють пороки серця.
- МРТ та УЗД нирок.
- Рентгенографічне дослідження опорно-рухового апарату.
- УЗД статевих органів.
Причини виникнення синдрому Шерешевського-Тернера
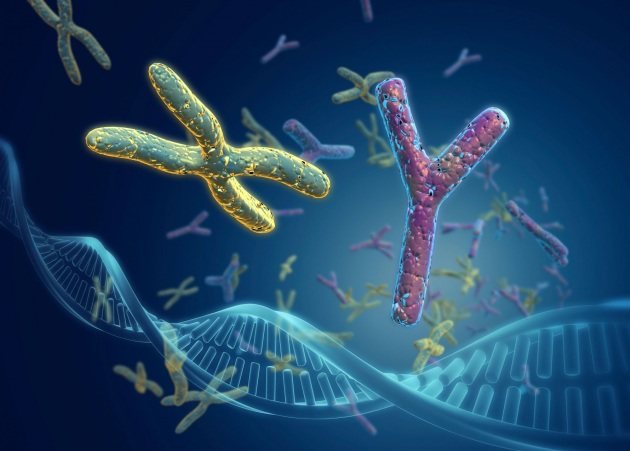
Нормальний людський каріотип відображається специфічної формулою і має вигляд 46, XY (у чоловіків) і 46, ХХ (у жінок). Цифра позначає число молекул ДНК, а букви – стать людини.
При синдромі Ульріха-Тернера молекул тільки 45, а однієї хромосоми немає. Дослідження, що дозволяє діагностувати недугу – кариотипирование.
Точна причина порушення невідома.
Етіологія
Основна причина формування синдрому Шерешевського-Тернера – це порушення нормального каріотипу, при якому спостерігається відсутність другої статевої хромосоми. Механізм передачі захворювання до кінця не з’ясований. У медичній сфері існує безліч суперечок про його спадковості. Факторами виникнення хвороби є:
- різні інфекції статевої системи, перенесені раніше майбутньою матір’ю;
- несприятливі умови навколишнього середовища, загазованість, забрудненість;
- вживання жінкою в великих кількостях спиртних напоїв під час вагітності;
- сильне випромінювання електромагнітного або іонізуючого характеру;
- голодування або будь-яке інше виснаження організму, наприклад, при важкої хвороби (найчастіше в період перед зачаттям).
У більшості випадків поява синдрому спостерігається випадково – часто такі діти народжуються у абсолютно здорових батьків. Це означає, що неможливо передбачити зачаття хворої дитини або заздалегідь провести профілактичні заходи. Під час вагітності можна дізнатися про хвороби тільки за допомогою аналізу на каріотип.
Основні симптоми
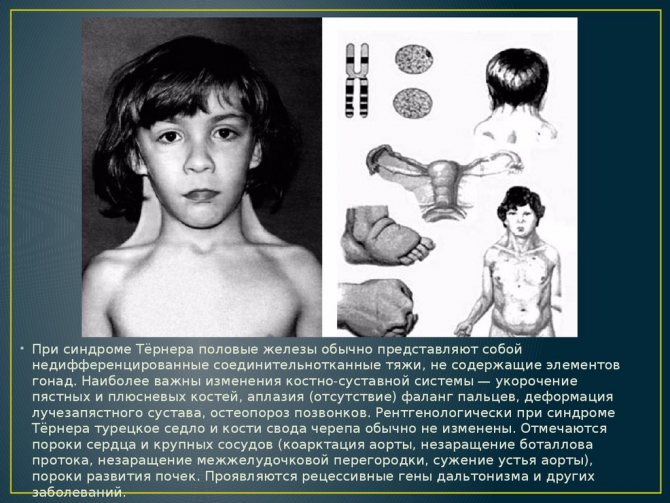
Виразність симптомів синдрому Ульріха-Тернера залежить від варіанту порушення. Головними ознаками недуги є кілька патологій.
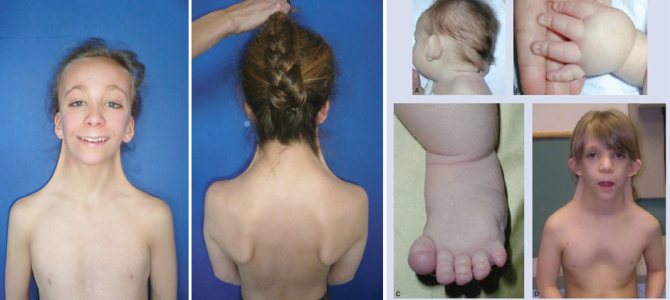
Шкірна складка на шиї
Аномалія вважається одним частих проявів захворювання (в 70% випадків). Вона має вигляд ущільнення крилоподібні форми, яке проходить від потилиці до трапецієподібним м’язам.
При значному надлишку шкіри проявляється видима, стягнута між головою і плечима, перетинка. Подібний косметичний дефект виправляється за допомогою нескладної операції.
Невеликий ріст
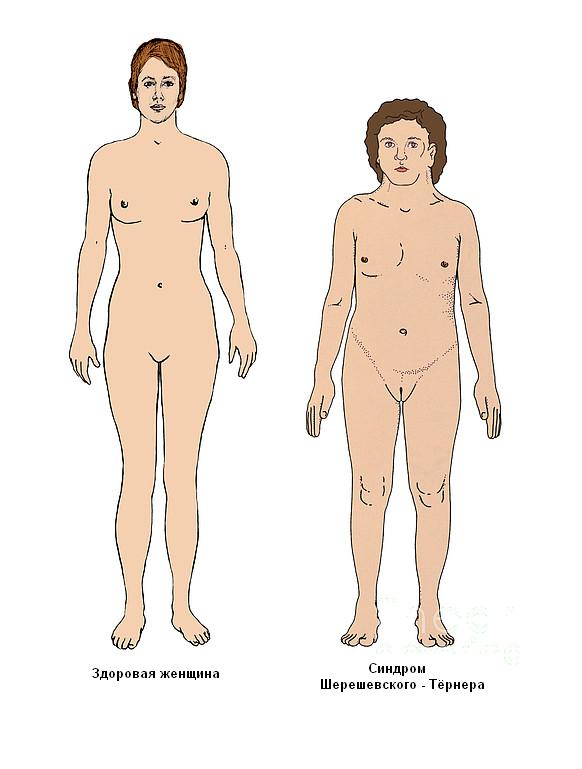
У новонароджених з генетичним порушенням довжина тіла варіюється в межах 42-45 см. Однак при мозаїчній формі аномалії зростання може бути нормальним. Розвиток відбувається з відставанням за жіночим типом.
Іноді низьке зростання обумовлюється порушенням у формуванні хребетного стовпа (зрощенням хребців або деформацією).
«Обличчя сфінкса»
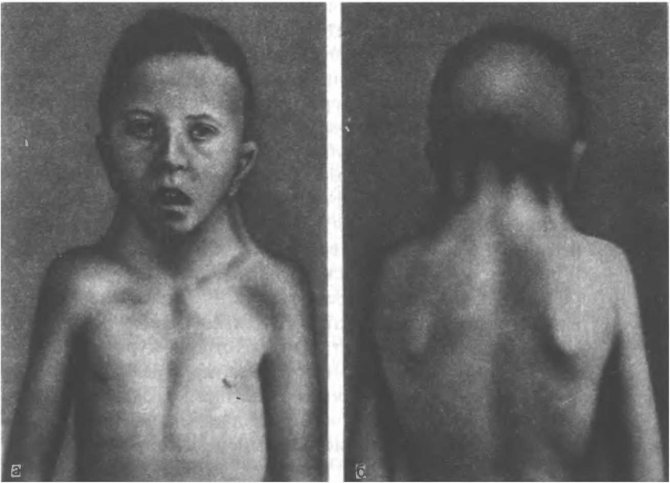
Ознака може виникати і при інших захворюваннях, однак при синдромі Ульріха-Тернера діагностується в 35% випадків і доповнюється шийної шкірної патологією.
Симптоми і ознаки
У новонароджених з синдромом Тернера-Шерешевського спостерігаються такі симптоми:
- низький зріст і вагу при народженні в термін;
- крилоподібні шкірні освіти на короткій шиї;
- виражена набряклість стоп і гомілок.
У віці до трьох років характерні такі прояви захворювання:
- надлишкова рухова активність;
- поганий апетит;
- затримка психомоторного розвитку;
- уповільнення темпів зростання;
- розумова відсталість (в 30% випадків);
- деформація вух, ліктьових суглобів, вкорочення п’ясткових кісток.
У період статевого дозрівання приєднується ряд ознак:
- зріст нижче середнього (130-145 см);
- широка грудна клітка;
- часті переломи внаслідок розрідження кісткової речовини;
- викривлення хребта (сколіоз);
- множинні пігментні плями на шкірі (невуси);
- надмірне оволосіння;
- нерозвинена тканину молочних залоз;
- відсутність менструації (аменорея).
У дорослих жінок спостерігається безпліддя (неможливість настання вагітності).
типи порушень
Існує кілька типів захворювання: мозаїчний, з відсутністю однієї з статевих хромосом (повна моносомія) або змінами її структури. При повній моносомии у плода після 12 тижнів відбувається нормальний розвиток яєчників. Однак потім починається заміщення фолікулів сполучною тканиною, розвиваються різні пороки розвитку.
Повна відсутність однієї з статевих хромосом вважається найважчим типом синдрому. При ньому ознаки патології яскраво виражені і насилу коригуються.
При мозаїчному типі порушення симптоми неспецифічні, добре піддаються терапії, і рідко викликають ускладнення.
При структурні зміни каріотип нормальний, але одна хромосома значно пошкоджена. Ознаки специфічної зовнішності маловиражен.
етіопатогенетичні чинники
Синдром Шершевского-Тернера – спадкова патологія, обумовлена відсутністю статевої хромосоми, яка, можна сказати, остаточно робить з жінки жінку. Некоректне поділ клітин в процесі зачаття призводить до вибудовування генетичного матеріалу з явними порушеннями. До вказаного хромосомному дисбалансу ведуть неправильно «встали» молекули ДНК.
Недуга також розвивається в результаті неправильного формування Х-хромосоми. Причинами її аномального будови є:
- втрата ділянки хромосоми в результаті її розриву,
- переніс ділянки хромосоми,
- освіту хромосоми у вигляді кільця,
- інші хромосомні перебудови – мутації або аберації.
Мозаїцизм має велике значення в розвитку недуги. У хворих в тканинах виявляють генетично різнорідні клітини в різних варіаціях. Всі ці варіанти характерні для жінок. У чоловіків синдром виникає вкрай рідко. Його основними причинами є транслокація або мозаїцизм. Мейотіческое розбіжність хромосом лежить в основі патологічного процесу.
Некоректна хромосомная «збірка» може полягати в наявності Y-хромосомного елемента в каріотипі. Таким хворим видаляють яєчники. Це необхідний захід, що дозволяє продовжити життя людям з даними недугою, оскільки Y-хромосомний елемент часто провокує розвиток ракової пухлини – гонадобластома.
Каріотипи при даному синдромі:
- Каріотип 45Х0 – заміна залозистої тканини яєчника сполучнотканинними тяжами. Функціонуючі жіночі залози призводять до необоротного безпліддя. Для продовження роду вдаються до допомоги ЕКО. Цей тип синдрому є найпоширенішим і одним з найважчих. Він відрізняється яскраво вираженою симптоматикою і розвитком важких ускладнень. Синдром насилу піддається лікуванню.
- Мозаїчна каріотип 45 X0 / 46 XY – відсутність матки і недорозвинення піхви, високий ризик онкології. Для попередження рецидивів хвороби показано видалення яєчників. Мозаїчна каріотип 45 Х0 / 46 ХХ – мізерно малі розміри яєчників. Вагітність можлива за участю донорської яйцеклітини. Мозаїчна тип синдрому відрізняється більш легким перебігом: вади розвитку не спостерігається, симптомів виникає набагато менше і вони менш виражені. Недуга добре лікується. Мозаїчна тип відрізняється поєднанням двох видів клітин – з нормальним каріотипом і без однієї Х-хромосоми. Від їх пропорційного співвідношення буде залежати стан здоров’я жінки.
В даний час вчені встановили, що синдром виникає спонтанно. Вік, спадковість, спосіб життя батьків і їх шкідливі звички не роблять істотного впливу на формування аномалії. Можливо деформація хромосоми відбувається під час запліднення під впливом патогенних факторів – іонізуючого або рентгенівського випромінювання, загазованості і забруднення навколишнього середовища, сильного електромагнітного впливу.
Спочатку у ембріона закладає нормальна кількість статевих клітин. У процесі росту і розвитку плода вони піддаються інволюції. Новонароджена дівчинка має дуже мало фолікулів в яєчнику або не має їх зовсім. Крім дисфункції яєчників у хворих в процесі ембріогенезу формуються численні вади внутрішніх органів.
діагностика
При вагітності запідозрити недугу можна під час виконання УЗД. При синдромі Шерешевського-Тернера з’являються деякі відхилення:
- потовщення комірцевої зони;
- шийна гігрома;
- розширення ниркових мисок і чашечок;
- деформація кісток, форми голови, кінцівок;
- серцеві вади;
- маловоддя або багатоводдя.
ТВП і гігрома помітні вже на першому плановому УЗД. При мозаїчному типі патології результати ультразвукової діагностики залишаються нормальними.
Для уточнення форми порушення рекомендується кариотипирование плода. Дослідження проводиться з 10 до 12 тижнів вагітності.
При здійсненні цієї процедури (під контролем УЗД) спеціальною голкою (вводиться в маткову порожнину через передню черевну стінку) або катетером (через шийку матки), виконують забір навколоплідної рідини (амніоцентез), крові з судин пуповини (кордоцентез) або зразка клітин плоду (біопсія хоріона).
Існує невеликий ризик викидня (1-2% випадків).
Для уточнення діагнозу після лікарського огляду знадобиться виконання деяких досліджень і діагностичних процедур:
- аналіз крові на концентрацію статевих гормонів;
- денситометрія;
- кариотипирование (якщо не виконувалося при вагітності);
- УЗД черевної порожнини, серця, нирок, органів малого таза;
- рентгенографія кистей, ліктьових суглобів, хребта.
Виконується диференціальна діагностика з синдромом Ульріха-Нунана, гіпофізарний гипогонадизмом, синдромом Майєра-Рокітанского-Кюстнера.
симптоматика
Патологія проявляється наступними симптомами:
- Новонароджені мають масу тіла від 2,5 кг до 2,8 кг і довжину тіла – менше 42-48 см.
- Шкірні складки з боків короткої шиї.
- Лимфедема кистей і ступень з наростанням лімфатичного набряку м’яких тканин.
- Деформовані нігтьові пластини.
- Порушення смоктального рефлексу, часті відрижки, блювота.
- Психомоторне збудження.
- Порушення мови, уваги і пам’яті.
- Рецидивуюче запалення середнього вуха, формування кондуктивної приглухуватості.
- Низький зріст дітей.
- Неправильна статура.
- Невиразна міміка, відсутність складок на лобі, потовщена і отвисшая нижня губа, напіввідкритий рот, деформовані вуха, низький ріс волосся, незвичайна форма грудної клітини, микрогнатия і мікрогенія.
- Атипично сформовані в процесі ембріогенезу або постнатальному періоді тазостегнові і ліктьові суглоби, укорочені кістки зап’ястя, неправильний контур ніг в результаті «О» – або «Х» -образної деформації гомілки, вкорочення пальців рук, сколіоз.
- Часті переломи внаслідок остеопорозу.
- «Готичне» небо, високий тембр голосу, аномалії зубів, неправильний прикус.
- Інтелект у хворих повністю зберігається. Діти легко засвоюють шкільну програму, активно проводять своє дозвілля, ведуть повноцінне життя.
- Психічний статус – інфантильність і ейфорія. Психічні порушення проявляються емоційною лабільністю, депресією, неврозами, тривогою і занепокоєнням.
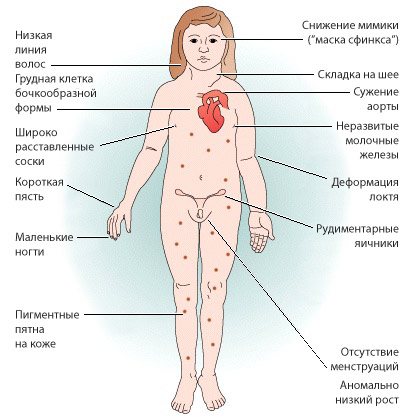
Статеве недорозвинення відрізняється певною своєрідністю. Всі жінки з синдромом Тернера страждають гіпогонадизмом – недорозвиненням яєчників. У них відсутні фолікули, а самі вони поступово заміщаються сполучнотканинними волокнами. Рудиментарна матка має малий розмір, великі статеві губи за формою нагадують мошонку, пліва і клітор недорозвинені, піхву лійкоподібної форми. Молочні залози мають апігментірованний, втягнутий, низькорозташованого сосок. На лобку і в пахвових западинах відзначається убогий зростання волосся або їх відсутність. Менструації часто затримуються або не наступають.
У хворих крім статевої дисфункції є вроджені аномалії внутрішніх органів:
- пороки серця і аорти;
- нефропатія зі стійкою артеріальною гіпертензією;
- опущення верхньої повіки, наявність епікантуса, дальтонізм, міопія;
- численні родимки і пігментні плями на тілі;
- надмірний ріст волосся;
- геродермія – патологічна атрофія шкіри, що нагадує старечу;
- цукровий діабет, ціліакія, ожиріння, гіпотиреоз;
- розширення дрібних судин травного тракту, внутрішні кровотечі.
Не у всіх дітей клінічні ознаки з’являються в повному обсязі. У різних жінок синдром не може проявлятися однаково. Цим він і примітний. Неможливо знайти навіть кілька ідентичних випадків хвороби.
Деякі діти з даним захворюванням народжуються без видимих зовнішніх ознак. Діагноз синдрому зазвичай їм ставлять в 12-14 років, коли дівчатка приходять на прийом до гінеколога. При відсутності своєчасного лікування в організмі розвиваються незворотні зміни. Для цієї недуги дуже важлива рання діагностика.
прогноз
Прогноз при синдромі Тернера сприятливий щодо тривалості життя і інтелектуального розвитку. Виняток становлять випадки виникнення:
- ВПС;
- розщеплення хребта;
- аномального формування сечостатевої системи.
Що стосується відновлення дітородної функції, то в основному пацієнтки залишаються безплідними. Однак при адекватної та своєчасної терапії жінки можуть створювати сім’ї, вести повноцінне сексуальне життя, народжувати дітей.
Синдром Шерешевського-Тернера – складна генетична патологія, що впливає на весь організм. Однак при своєчасній діагностиці і правильно виконаному лікуванні пацієнтки мають шанс адаптуватися в соціумі.
лікування
Основними завданнями лікування пацієнток з синдромом Шерешевського-Тернера служать стимуляція зростання, індукція формування вторинних статевих ознак і регулярного менструального циклу. У ранньому дитинстві лікування неспецифічної – масаж, ЛФК, вітаміни, повноцінне харчування, охоронний режим.
З метою збільшення кінцевого зростання призначається рекомбінантний гормон росту (соматотропін) у вигляді щоденних підшкірних ін’єкцій до досягнення пацієнткою кісткового віку 15 років і зменшення швидкості росту до 2 см в рік. У більшості випадків ростостимулюючі терапія допомагає хворим вирости до 150-155 см. Лікування гормоном росту рекомендується поєднувати терапією анаболічними стероїдами.
Для імітації нормального статевого дозрівання з 13-14 років призначається замісна терапія естрогенами, а через 12-18 місяців циклічна терапія естроген-прогестагенну оральними контрацептивами. Замісна гормонотерапія проводиться до віку природної менопаузи у здорових жінок (приблизно до 50 років). Чоловікам з синдромом Шерешевського-Тернера призначається ЗГТ чоловічими статевими гормонами.
При гемодинамічно значущих ВПС здійснюється їх хірургічна корекція. Усунення крилоподібних складок шиї проводиться методами пластичної хірургії.
При досягненні адекватного рівня статевого розвитку жінки з синдромом Шерешевського-Тернера можуть мати дітей за допомогою ЕКЗ, використовуючи донорську яйцеклітину. При наявності короткочасної овариальной активності можливе використання для запліднення власних ооцитів. Проблема надмірного росту волосся вирішується за допомогою епіляції.
Синдром Мея-Тернера і варикозна хвороба вен органів малого таза у чоловіків
А.А. Капто ФГАОУ ВО «Український університет дружби народів»; Україна, 117198 Київ, вул. Миклухо-Маклая, 6
Вступ
Компресія лівої загальної клубової вени і фіброзні спайки в ній вперше були описані німецьким патологом R. Virchow в 1851 р [1]. Він зазначив, що глибокі ілеофеморальний тромбози зустрічалися в 5 разів частіше в лівій нозі, ніж у правій. За даними дослідників [2-5], компресія лівої загальної клубової вени – широко поширена патологія, яка зустрічається у 22-50% населення (без поділу за статевою ознакою) (табл. 1).
Таблиця 1. Частота виявлення фіброзних спайок в просвіті лівої загальної клубової вени при аутопсії (без поділу за статевою ознакою)
| Автор | рік публікації | Кількість аутопсії, абс. | Частота виявлення спайок,% |
| JP McMurrich [2] | 1908 | 57 | 30 |
| WE Ehrich, EB Krumbhaar [3] | 1943 | 412 | 23,8 |
| R. May, J. Thurner [4] | 1957 | 430 | 22 |
| N. Usui і співавт. [5] | 1978 | 90 | 50 |
В англомовній літературі синдром компресії лівої загальної клубової вени частіше називають синдромом Мея-Тернера (May-Thurner syndrome). Безсимптомна компресія лівої загальної клубової вени у дорослого населення, за даними HC Baron і співавт., Зустрічається в 16-20% випадків [6]. Природний перебіг синдрому Мея-Тернера є передумовою розвитку ілеофеморальний тромбозу і посттромботичного синдрому. Розгорнута клінічна картина ілеофеморальний тромбозу у хворих з синдромом компресії лівої загальної клубової вени також називається синдромом Кокетта (Cockett syndrome) [7, 8].
До недавнього часу актуальність проблеми синдрому Мея-Тернера пов’язували тільки з його тромботическими ускладненнями. При нетромботіческій компресії лівої загальної клубової вени розвивається клінічна картина, обумовлена полнокровием як внутрішньої, так і зовнішньої клубових вен. Повнокров’я внутрішньої клубової вени обумовлює повнокров’я вен органів малого таза через розвиток колатерального кровообігу. Повнокров’я зовнішньої клубової вени призводить до появи ретроградного кровотоку під впадає в неї лівої кремастерний вені та формування ілеосперматіческого типу варикоцеле.
Вивчення артеріовенозних конфліктів як причини варикозної хвороби вен органів малого таза у чоловіків почалося порівняно недавно. Гіпотеза про роль венозного повнокров’я органів малого таза в розвитку різних урологічних захворювань була висловлена ще в 1895 році німецьким урологом K. Posner в його монографії «Діагностика сечостатевих хвороб» [9]. В.В. Яковенко (1955) вперше припустив, що причиною розвитку варикоцеле є венозний застій в сечостатевому сплетінні [10]. H. Sakamoto і Y. Ogawa (2008) досліджували взаємозв’язок між варикоцеле і венозних сплетінням передміхурової залози (ПЖ) за допомогою скротальной доплерографії і трансперінеальной кольорової доплерографії. У всіх 209 чоловіків, включених в дослідження, діаметр вен простатичного сплетіння (ПС) позитивно корелював з діаметром вен правого і лівого гроздевідних сплетінь. Таким чином автори встановили, що варикоцеле, особливо двостороннє, пов’язане з венозними аномаліями ПЖ [11]. А.І. Неймарк та співавт. (2013) запропонували виділяти 2 види варикоцеле: 1) ізольоване, при якому відзначаються мінімальні порушення гемодинаміки в правому насінники, а ПЖ не залучена в патологічний процес; 2) поєднується з тазової конгестии, при якому порушення зачіпають не тільки ліве, а й праве яєчко, а також ПЖ [12]. Критеріями варикозної хвороби малого тазу у чоловіків А.Ю. Цуканов і Р.В. Ляшев (2014 року) вважали діаметр вен парапростатіческой сплетення> 5 мм і / або наявність ретроградного кровотоку при пробі Вальсальви, реєстрованого при дуплексному скануванні вен з використанням ректального датчика [13]. А.А. Капто, О.Б. Жуков (2016) представили перший в світі огляд літератури по варикозної хвороби вен органів малого таза у чоловіків [14]. У 2017 р нами запропонована ультразвукова класифікація варикозного розширення вен ПС як маркера варикозної хвороби вен органів малого таза у чоловіків [15].
В даний час не існує ні приватних, ні загальних рекомендацій з діагностики та лікування тазової венозної конгестии і ілеофеморальний судинних конфліктів у чоловіків з урологічної і андрологічної патологією. У зв’язку з цим мета цього дослідження – вивчення методів діагностики і лікування клубової венозної компресії у чоловіків з урологічної і андрологічної патологією і варикозну хворобу вен органів малого таза.
матеріали та методи
З 2019 по 2019 р обстежені 110 пацієнтів у віці від 17 до 69 років (в середньому 33,2 року) з двостороннім варикоцеле, варикозну хворобу вен органів малого таза і синдромом Мея-Тернера.
Діагноз варикоцеле був верифікований за даними огляду та ультразвукового дослідження (УЗД) з доплерографією органів мошонки. Як ультразвукового критерію постановки діагнозу варикоцеле ми прийняли діаметр вен гроздевидного сплетення> 2 мм в спокої в кліностазе, що відображає загальноприйняту точку зору [16-20].
Діагноз варикозної хвороби вен органів малого таза був верифікований за допомогою трансректального УЗД (ТРУЗІ) з використанням критеріїв і класифікації, запропонованої нами в 2017 р (табл. 2) [21].
Таблиця 2. Ультразвукова класифікація варикозного розширення вен передміхурової залози по А.А. Капто [21]
| стадія | визначення варикозу | Максимальний діаметр вен, мм | Швидкість кровотоку, см / с | Швидкість кровотоку при пробі Вальсальви, см / с |
| I | Відомий | <4 | <3 | <5 |
| II | Значний | 5-10 | 3-5 | 5-15 |
| III | виражений | > 10 | > 5 | > 15 |
Синдром Мея-Тернера діагностували за допомогою магнітно-резонансної томографії (МРТ) вен з контрастуванням. Дослідження проведено на магнітно-резонансному томографі GE Opima MR360 (General Electric, США). Для діагностики аортомезентеріальной компресії визначали величину аортомезентеріального кута (aortomesenteric angle) (норма 28-65 °) і аортомезентеріальное відстань (aortomesenteric distance) (норма 10-34 мм) [22, 23], для діагностики синдрому Мея-Тернера – величину нижнього кута поперекового лордозу (lower lumbar lordosis angle) (норма> 134,3 °) і діаметр тунелю клубової вени (норма> 4,2 мм). [24]. Варіанти артеріовенозних конфліктів ілеокавальном сегмента оцінювали за допомогою запропонованої нами в 2019 р класифікації (рис. 1) [25].
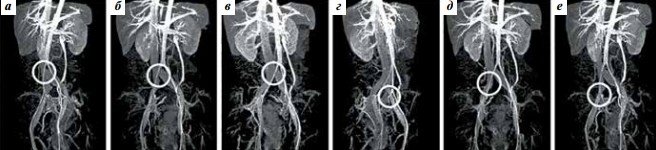
Рис. 1. Топографо-анатомічна класифікація варіантів компресії клубових вен по А.А. Капто [25]. Магнітно-резонансне дослідження нижньої порожнистої вени і судин малого таза (моделювання): а – центральний проксимальний (висока біфуркація аорти, при якій права загальна клубова артерія здавлює нижній відділ нижньої порожнистої вени до її поділу на клубові вени); б – центральний дистальний (висока біфуркація аорти, при якій права загальна клубова артерія здавлює нижній відділ нижньої порожнистої вени в місці її поділу на клубові вени); в – лівий проксимальний (права загальна клубова артерія здавлює ліву загальну клубову вену (синдром Мея-Тернера)); г – лівий дистальний (компресія лівої зовнішньої і / або лівої внутрішньої клубової артерією лівої зовнішньої клубової вени); д – правий проксимальний (компресія правої загальної клубової артерією правої загальної клубової вени); е – правий дистальний (компресія правої зовнішньої і / або правої внутрішньої клубової артерією правої зовнішньої клубової вени)
Для діагностики порушень ерекції застосовували міжнародний індекс еректильної функції (International Index of Erectile Function, МІЕФ-5) [26], проводили доплерографію судин статевого члена в стані спокою і при фармакологічно індукованої ерекції. При підозрі на веногенную еректильну дисфункцію виконували динамічну фармакокавернозографія, яка дозволяла верифікувати проксимальний, дистальний і змішаний типи патологічного венозного відтоку з кавернозних тіл статевого члена.
У пацієнтів з урологічними і андрологічними захворюваннями абсолютним показанням до ендоваскулярної операції було поєднання таких ознак клубової венозної компресії:
- виражених симптомів з боку тазових органів (болі, дизурії, еректильної дисфункції);
- двостороннього та / або рецидивного варикоцеле; 3) варикозного розширення вен ПЖ II-III ступеня (максимальний діаметр вен ПС> 5 мм);
- компресії клубових вен за даними МРТ та флебографії;
- наявність колатеральних гілок клубових вен за даними флебографії.
Ізольована патоспермії без інших клінічних проявів зустрічалася вкрай рідко (n = 4) і не розглядалася як абсолютне показання до ангіопластики і стентування клубових вен.
Оперативне лікування синдрому Мея-Тернера проводилося під місцевою анестезією в умовах рентгеноoпераціонной і включало наступні етапи:
- пункцію стегнової або підколінної вени (під ультразвуковим контролем);
- мультіпроекціонную интраоперационную флебографию;
- балонну ангіопластику;
- імплантацію стента;
- постділатацію стентоване сегмента;
- контрольну флебографию.
Перед- і післяопераційне ведення пацієнта включало:
- антикоагулянтну терапію ривароксабаном в дозі 20 мг / добу протягом 1 тижня до операції і 6 міс після неї;
- УЗД клубових судин в 1-у добу після операції, через 2 тижні і через 1, 3, 6 міс;
- УЗД органів мошонки з кольоровим доплерівським картуванням до операції і через 1, 3, 6, 9 і 12 міс після неї;
- ТРУЗІ ПЖ і вен ПС до операції і через 1, 3, 6, 9 і 12 міс після неї;
- оцінку стану пацієнта за допомогою МІЕФ-5, міжнародної шкали тяжкості симптомів при захворюваннях ПЖ (International Prostatic Symptom Score, IPSS) [27], індексу симптомів хронічного простатиту Національних інститутів охорони здоров’я США (National Institutes of Health Chronic Prostatitis Symptom Index, NIH-CPSI) [28] до операції і через 1, 3, 6, 9 і 12 міс після неї.
Статистичну обробку результатів провели за допомогою статистичних пакетів Microsoft Office, програми Excel. Статистичну значимість відмінностей показників, що мають нормальний розподіл, оцінювали за критерієм Стьюдента, U-критерію Манна-Уїтні.
результати
Вік 110 пацієнтів, включених у дослідження, варіював від 17 до 69 років і в середньому становив 33,2 року.
Причиною звернення пацієнтів були болі (внизу живота, в області зовнішніх статевих органів, внизу попереку), дизурія (іррітатівний і обструктивна симптоматика), еректильна дисфункція (погіршення ранкових, спонтанних і адекватних ерекцій), патоспермії (оліго-, астено і тератозооспермия), піоспермія (як маркер запального процесу в ПЖ), рецидивний і не піддається лікуванню хронічний простатит, двостороннє та рецидивної варикоцеле. Найчастіше пацієнти пред’являли скарги на болі. Із супутніх хірургічних патологій в більшості випадків зустрічалися геморой і варикозне розширення вен нижніх кінцівок.
Варікоцелектомія в анамнезі була у 39 пацієнтів: 1 операція – у 23, 2 – у 9, 3 – у 2, 4 – у 1, 5 – у 1. З них 15 пацієнтів були раніше прооперовані нами.
При УЗД органів мошонки у всіх випадках верифікувати двостороннє варикоцеле, більш виражене з лівого боку
За даними ТРУЗІ, обсяг ПЖ варіював від 6,6 до 76,6 см3 і в середньому становив 21,2 см3. Кісти ПЖ були виявлені у 2 пацієнтів. Збільшена середня частка ПЖ була виявлена у 18 пацієнтів (у віці від 20 до 55 років, в середньому 36,3 року). Обсяг ПЖ у пацієнтів зі збільшеною середньою часткою варіював від 9,3 до 37,9 см3 і в середньому становив 23,3 см3 (рис. 2).
Рис. 2. Трансректальне обстеження передміхурової залози пацієнта Х., 39 років, з синдромом Мея-Тернера, рецидивних двостороннім варикоцеле (3 операції в анамнезі), варикозну хворобу вен органів малого таза III стадії, збільшеною середньою часткою передміхурової залози при її обсязі 22,2 см3 і вираженою дизурією (іррітатівной і обструктивної симптоматикою). За даними урофлоуметріі, середня швидкість потоку сечі склала 6,2 см / с
Відня ПС при ТРУЗІ візуалізовані у всіх пацієнтів. Максимальний діаметр вен ПС праворуч – від 2,3 до 14,1 мм, в середньому 7,1 мм. Максимальний діаметр вен ПС зліва – від 5,0 до 17,1 мм, в середньому 9,7 мм. При доплерографії судин ПЖ при пробі Вальсальви відзначалося збільшення швидкості кровотоку по венах з 5 до 15 см / с. Таким чином, у всіх пацієнтів виявлено білатерально розширення вен ПС, що вказувало на двосторонній характер варикоцеле. Переважання лівостороннього варикоцеле над правостороннім позитивно корелювало з переважанням максимального діаметра вен ПС зліва над максимальним діаметром вен справа.
МРТ нижньої порожнистої вени і судин малого таза і верифікація клубової венозної компресії проведені у 110 пацієнтів. Аналіз отриманих даних дозволив нам виділити 4 стадії розвитку (або форми) синдрому Мея-Тернера (рис. 3).
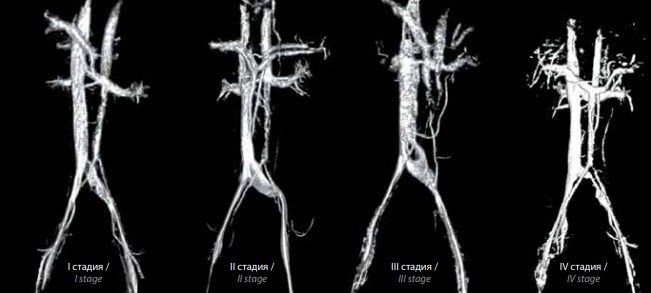
Рис. 3. Стадії синдрому Мея-Тернера за даними магнітно-резонансної томографії нижньої порожнистої вени і судин малого таза з 3D-реконструкцією: I – компресія лівої загальної клубової вени; II – компресія лівої загальної клубової вени з її дилатацією; III – компресія лівої клубової вени зі змиканням стінок посудини в центральній його частині і з її дилатацією; IV – виражене і протяжне звуження просвіту лівої загальної клубової вени
Артеріовенозний конфлікт діагностували також з використанням значень аортомезентеріального кута, аортомезентеріального відстані, нижнього кута поперекового лордозу і діаметра тунелю клубової вени (табл. 3).
Таблиця 3. Показники артеріовенозного конфлікту за даними магнітно-резонансної томографії вен, n = 110
| показник | мінімум | максимум | середнє | норма |
| Аортомезентеріальний кут, градуси | 9,6 | 120,6 | 43,6 | 28-65 |
| Аортомезентеріальное відстань, мм | 2,5 | 49,2 | 13,7 | 10-34 |
| Нижній кут поперекового лордозу, градуси | 112,2 | 134,3 | 123,9 | 134,33- 136,76 |
| Діаметр тунелю клубової вени, мм | 0,9 | 5,1 | 2,5 | 4,18- 4,50 |
За даними МРТ вен аортомезентеріальная компресія (синдром аортомезентеріального пінцета, синдром горіхоколи, nutcracker syndrome) в поєднанні з синдромом Мея-Тернера була діагностована у 32 (29,1%) пацієнтів. Ретроаортальная ліва ниркова вена (задній синдром горіхоколи, posterior nutcracker syndrome) в поєднанні з синдромом Мея-Тернера була виявлена в 4 (3,6%) випадках. Здавлення клубової вени без поєднання з аортомезентеріальной компресією спостерігалося у 74 (67,3%) пацієнтів. Центральний проксимальний артеріовенозний конфлікт був виявлений у 4 пацієнтів, центральний дистальний – у 7, лівий проксимальний – у 90, лівий дистальний – у 43, правий проксимальний – у 3, правий дистальний – у 5 пацієнтів. Різні поєднання артеріовенозних конфліктів клубових судин були виявлені в 43 (39,1%) випадках. Аналіз отриманих даних дозволив нам відзначити, що той чи інший конфлікт діагностований у 70 (63,6%) пацієнтів, а їх поєднані форми – у 40 (36,4%) (табл. 4).
Таблиця 4. Поширеність різних типів артеріовенозних клубових конфліктів, n = 110
| Тип конфлікту | число випадків | |
| абс. | % | |
| Лівий проксимальний, або синдром Мея- Тернера | 56 | 51 |
| лівий дистальний | 6 | 5 |
| Лівий проксимальний + лівий дистальний | 32 | 29 |
| Лівий проксимальний + правий проксимальний | 2 | 2 |
| Лівий проксимальний + правий дистальний | 1 | 1 |
| центральний проксимальний | 3 | 3 |
| центральний дистальний | 2 | 2 |
| Центральний дистальний + лівий дистальний | 4 | 3 |
| правий дистальний | 3 | 3 |
| Центральний дистальний + лівий дистальний + правий дистальний | 1 | 1 |
| всього | 110 | 100 |
Скарги на недостатню ерекцію пред’являли 78 (70,9%) з 110 пацієнтів. Доплерографію судин статевого члена в стані спокою і при фармакологічно індукованої ерекції виконали у 48 пацієнтів. У 40 випадках верифікувати веногенную еректильну дисфункцію. Динамічну фармакокавернозографія провели у 22 пацієнтів; патологічний венозний дренаж виявили у 18: проксимального типу – у 10, дистального – у 2, змішаного – у 6.
Флебографію як комплексне ангіографічне дослідження для виявлення артеріовенозних конфліктів верхнього (синдрому аортомезентеріального пінцета) і нижнього (синдрому Мея-Тернера) рівнів виконали у 41 пацієнта. Вона включала в себе ренокаваграфію і ілєокаваграфію з флеботонометра. У всіх випадках було виявлено синдром Мея-Тернера, а в 12 випадках він поєднувався з синдромом аортомезентеріального пінцета.
Аналіз отриманих при флебографії ілеокавальном сегмента даних дозволив нам виділити 4 стадії синдрому Мея-Тернера в залежності від наявності і вираженості колатерального кровообігу (рис. 4).
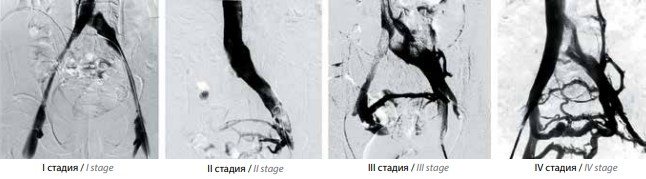
Рис. 4. Стадії синдрому Мея-Тернера за даними ілєокаваграфію в залежності від наявності і вираженості колатерального кровообігу: I – відсутність контрастування вен таза; II – контрастування вен таза; III – контрастування вен таза з перетіканням контрасту в контралатеральную праву загальну клубову вену; IV – контрастування вен таза з перетіканням контрасту в праву загальну клубову вену і висхідні поперекові вени зліва
Ми прийшли до висновку, що підвищення тиску в загальній клубової вені при виконанні проби Вальсальви більш ніж в 3 рази може вказувати на наявність клубової венозної компресії (табл. 5).
Оперативне лікування синдрому клубової венозної компресії здійснили у 26 пацієнтів. З огляду на те, що фіброзні спайки в здавленої клубової вени зустрічаються в більшості випадків, будучи неминучим патогенетичним ланкою цього захворювання, проведення ангіопластики перед стентуванням ми вважали обов’язковим (рис. 5). Для балонної ангіопластики найчастіше ми використовували дилатаційних катетери Atlas Gold PTA Dilatation Catheter (Bard Peripheral Vascular).
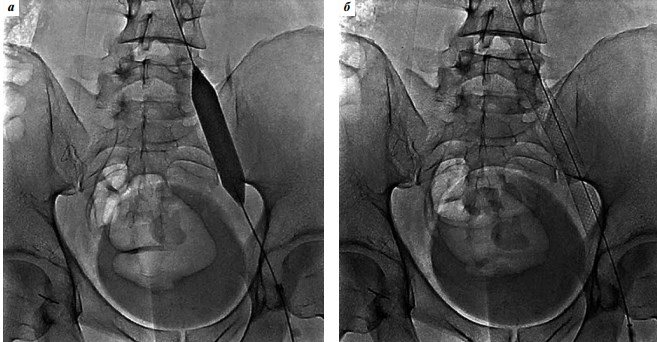
Рис. 5. Балонна ангіопластика (а) і стентування (б) лівої загальної клубової вени при оперативному лікуванні синдрому Мея-Тернера
Таблиця 5. Результати флеботонометра у пацієнтів з клубової венозної компресією, n = 25
| Відень | Тиск, мм рт. ст | Кратність збільшення тиску | |
| в спокої | при пробі Вальсальви | ||
| Нижня порожниста | 5,25 | 10,35 | 1,97 |
| Права загальна клубова | 6,22 | 15,25 | 2,45 |
| Ліва загальна клубова | 8,35 | 26,28 | 3,14 |
З нашої точки зору, балонна ангіопластика без інших методів неефективна, а імплантація стента обов’язкове. Для імплантації ми використовували венозні стенти Wallstent-Uni Endoprosthesis (Boston Scientific) зі сплаву Elgiloy (на основі нікелю, кобальту і хрому). Діаметр і довжину стента підбирали в ході операції. Стент імплантували таким чином, щоб його верхня частина щільно прилягала до стінок лівої загальної клубової вени, а його верхній край не виходив в просвіт нижньої порожнистої вени. Встановлювані венозні стенти є самораскривающіеся. Незважаючи на це, у всіх випадках оперативного лікування синдрому клубової венозної компресії після імплантації стента ми проводили постділатацію стентоване ділянки в проксимальному і дистальному відділах. Після стентування виконували контрольну флебографию і флеботонометра, які підтверджували відсутність компресії лівої загальної клубової вени, відсутність колатеральногокровообігу і кої клубової гіпертензії. Вбольшінстве випадків була потрібна імплантація 1 стента, навіть якщо подвздошная венозна компресія носила поєднаний характер (наприклад, лівий проксимальний + лівий дистальний конфлікти). Однак у 2 пацієнтів встановили по 2 стента: у 1 – в ліву загальну і ліву зовнішню клубову вени, у 1 – в ліву і в праву загальні клубові вени.
Найбільш виражені зміни скарг і об’єктивних даних спостерігалися до 3-го місяця після ангіопластики і стентування (рис. 6). Нами були отримані наступні середні значення: МІЕФ-5 до операції – 17,4, через 3 місяці після неї – 20,9; IPSS відповідно 21,7 і 15,3; NIH-CPSI – 28,4 і 22,3.
Через 3 місяці після ангіопластики і стентування у всіх випадках максимальний діаметр вен ПЖ зменшився на 20-70% (в середньому на 45,6%) за даними ТРУЗІ.
У 13 пацієнтів з ізольованою клубової компресією (без поєднання з синдромом аортомезентеріального пінцета) до 3-го місяця після ангіопластики і стентування відбувалася редукція варикоцеле: у всіх випадках діаметр лівої і правої яєчкові вен в кліностазе в спокійному стані став <2 мм.
У 4 пацієнтів з поєднанням синдрому аортомезентеріального пінцета і клубової венозної компресії через 6 місяців після ангіопластики і стентування лівої загальної клубової вени була виконана двостороння варікоцелектомія з серединного мошоночних доступу по лінії Веслінга з позитивним результатом.
Обговорення
BL Coolsaet в 1980 р за результатами ангіографічної обстеження 67 пацієнтів виділив 3 типи лівостороннього варикоцеле:
- рефлюкс з лівої ниркової вени у внутрішню яічковую вену (реносперматіческій тип);
- рефлюкс з лівої загальної клубової вени в екстрафунікулярние вени, що викликано обструкцією лівої загальної клубової вени, куди вони впадають (ілеосперматіческій тип);
- комбінація перших 2 типів (змішаний тип) [29].
Таким чином, BL Coolsaet вперше визначив роль клубової венозної компресії, або, в більш вузькому сенсі, синдрому Мея-Тернера, в розвитку варикоцеле. MD Bomalaski і співавт. в 1993 р вперше описали стійке до хірургічного лікування варикоцеле, розвинене внаслідок компресії лівої загальної клубової вени. Цей випадок продемонстрував необхідність відмови від стандартної хірургічної техніки в таких випадках [30].
Проведений раніше аналіз причин рецидивів варикоцеле у 14 (6,3%) з 223 пацієнтів, у яких протягом 10 років виконували операцію з трансскротального доступу по лінії Веслінга, дозволив нам верифікувати у всіх випадках синдром Мея- Тернера [31]. Разом з тим в сучасних клінічних посібниках України, Європи і Америки немає вказівок на те, що результативність традиційного хірургічного лікування може знизитися внаслідок синдрому Мея-Тернера.
Консервативних методів лікування синдрому Мея- Тернера не існує. CA Binkert і співавт. в 1998 р вперше опублікували звіт про успішну ангіопластики і стентування лівої загальної клубової вени при синдромі Мея-Тернера і тромботической хвороби у 8 пацієнтів (7 жінок і 1 чоловіка) [32]. З тих пір протягом 20 років рентгенохірургії проводять подібне лікування з приводу тромбозів і тромбоемболії легеневої артерії у пацієнтів з синдромом Мея- Тернера.
Незважаючи на те що роль клубової венозної компресії в розвитку варикоцеле була визначена ще в 1980 р BL Coolsaet, який запропонував гемодинамическую класифікацію, якою користується в даний час більшість урологів, до 2019 р ангіопластика і стентування у пацієнтів з урологічними і андрологічними захворюваннями не виконувалися. Перше в світі стентування лівої загальної клубової вени у пацієнтів з ілеосперматіческім типом варикоцеле здійснено нами 28.03.2017 в Києві [33]. Через 4 міс JR Stern і співавт. в Нью-Йорку виконали стентування у пацієнта з варикоцеле, розвинувся внаслідок синдрому Мея-Тернера [34]. На даний момент ми провели ангіопластику і стентування у 26 пацієнтів з синдромом Мея-Тернера, ілеосперматіческім типом варикоцеле і варикозну хворобу вен органів малого таза і отримали позитивні результати.
Тактика ведення пацієнтів з поєднанням синдрому аортомезентеріального пінцета і клубової венозної компресії включала наступні етапи:
- флебографию, ангіопластику і стентування лівої загальної клубової вени;
- оцінку стану скротального і пельвікального венозного повнокров’я через 6 міс з метою визначення показань до оперативного лікування варикоцеле.
У всіх випадках зменшення інтенсивності больового синдрому і його зникнення позитивно корелювали зі зниженням вираженості тазової венозної гіперволемії і зменшенням діаметра вен ПЖ. Це спостереження дозволяє під новим кутом розглянути проблему хронічного тазового болю і хронічного простатиту. Усунення венозного повнокров’я тазових органів в результаті ангіопластики і стентування лівої загальної клубової вени супроводжувалося поліпшенням або повним відновленням ерекції без застосування інгібіторів фосфодіестерази 5-го типу або проведення будь-якої іншої терапії. Це дозволило нам оцінити веногенную еректильну дисфункцію як складову частину проблеми варикозної хвороби вен органів малого таза у чоловіків.
висновок
У даній роботі представлений перший і найбільший в світі досвід оперативного лікування синдрому ілеофеморальний компресії як причини двостороннього варикоцеле (в тому числі рецидивного) і варикозної хвороби вен органів малого таза у чоловіків.
Аналіз отриманих даних дозволив нам запропонувати 2 нові класифікації: 1) 4 стадії розвитку синдрому Мея-Тернера за даними МРТ нижньої порожнистої вени і судин малого таза з 3D-реконструкцією; 2) 4 стадії компресії лівої загальної клубової вени при синдромі Мея-Тернера в залежності від наявності і вираженості колатерального кровообігу при флебографії ілеокавальном сегмента.
Отримані нами дані свідчать про те, що синдром Мея-Тернера може бути причиною рецидивного варикоцеле. Сучасний діагностичний алгоритм у пацієнтів з варикоцеле не орієнтований на виявлення синдрому Мея-Тернера. Показання до традиційного хірургічного лікування варикоцеле у пацієнтів з синдромом Мея-Тернера потребують перегляду.
До недавнього часу діагноз рецидивного варикоцеле внаслідок ілеофеморальний компресії, зокрема синдрому Мея-Тернера, був для урологів і андрологів «тупиковим» в плані лікування. Найбільш популярна останнім часом операція з JL Marmar і співавт. (Субінгвінальная варікоцелектомія) [35] не в змозі вирішити проблему ілеофеморальний компресії, вторинної тазової венозної гіперволемії, яка в цьому випадку з високим ступенем ймовірності стає рецидивної.
Ангіопластика і стентування клубових вен при артеріовенозних конфліктах – високоефективний метод лікування пацієнтів з варикозною хворобою вен органів малого таза в поєднанні з варикоцеле.
література
- Virchow R. Uber die Erweiterung Kleinerer Gefasse. Arch Path Anat 1851; 3 (3): 427-9. DOI: 10.1007 / BF01960918.
- McMurrich JP The occurrence of congenital adhesions in the common iliac veins and their relation to thrombosis of the femoral and iliac veins. Am J Med Sci 1908; 135: 342-5. DOI: 10.1097 / 00000441-190803000-00004.
- Ehrich WE, Krumbhaar EB A frequent obstructive anomaly of the mouth of the left common iliac vein. Am Heart J 1943; 26 (6): 737-50. DOI: 10.1016 / S0002-8703 (43) 90285-6.
- May R., Thurner J. The cause of the predominantly sinistral occurrence of thrombosis of the pelvic veins. Angiology 1957; 8 (5): 419-27. DOI: 10.1177 / 000331975700800505. PMID: 13478912.
- Usui N., Muraguchi K., Yamamoto H. et al. . Surgery 1978; 40: 983.
- Baron HC, Shams J., Wayne M. Iliac vein compression syndrome: a new method of treatment. Am Surg 2000; 66 (7): 653-5. PMID: 10917476.
- Cockett FB, Thomas ML The iliac compression syndrome. Br J Surg1965; 52 (10): 816-21. PMID: 5828716.
- Cockett FB, Thomas ML, Negus D. Iliac vein compression – its relation to iliofemoral thrombosis and the postthrombotic syndrome. Br Med J 1967; 2 (5543): 14-9. DOI: 10.1136 / bmj.2.5543.14. PMID: 6020994.
- Велика медична енциклопедія. Під. ред. Н.А. Семашко М .: Радянська енциклопедія, 1933. Т. 26. С. 70..
- Яковенко В.В. Венозні освіти яєчка, сім’яного канатика та хірургічне лікування варикоцеле. Реферат дис. … канд. мед. наук. Ленінград, 1955. 15 с. .
- Sakamoto H., Ogawa Y. Is varicocele associated with underlying venous abnormalities? Varicocele and the prostatic venous plexus. J Urol 2008; 180 (4): 1427-31. DOI: 10.1016 / j. juro.2008.06.048. PMID: 18710746.
- Неймарк А.І., Попов І.С., Газаматов А.В. Особливості мікроциркуляції передміхурової залози і гонад у юнаків, які страждають ізольованим варикоцеле і варикоцеле в поєднанні з тазової конгестии. Експериментальна та клінічна урологія 2013; (2): 56-60. .
- Цуканов А.Ю., Ляшев Р.В. Порушення венозного кровотоку як причина хронічного простатиту абактеріального (синдрому хронічної тазової болі). Урологія 2014; (4): 37-42. [Tsukanov A.Yu., Lyashev RV Disorders of venous blood flow as a cause of chronic abacterial prostatitis (chronic pelvic pain syndrome). Urologiya = Urology 2014; (4): 37-42. (In Russ.)].
- Капто А.А., Жуков О.Б. Варикозна хвороба малого тазу у чоловіків (огляд літератури). Андрологія і генітальна хірургія 2016 року; 17 (2): 10-9. .
- Капто А.А. Варикозне розширення вен передміхурової залози у пацієнтів з варикоцеле. Експериментальна та клінічна урологія 2017; (1): 98-103. .
- Wolverson MK, Houttuin E., Heiberg E. et al. High-resolution real-time sonography of scrotal varicocele. AJR Am J Roentgenol 1983; 141 (4): 775-9. DOI: 10.2214 / ajr.141.4.775. PMID: 6604430.
- Rifkin MD, Foy PM, Kurtz AB et al. The role of diagnostic ultrasonography in varicocele evaluation. J Ultrasound Med 1983; 2 (6): 271-5. https: // doi. org / 10.7863 / jum.1983.2.6.271. PMID: 6876259.
- Gonda RL Jr, Karo JJ, Forte RA, O’Donnell KT Diagnosis of subclinical varicocele in infertility. AJR Am J Roentgenol 1987; 148 (1): 71-5. https://dx.doi.org/10.2214/ajr.148.1.71. PMID: 3024475.
- Gerscovich EO High-resolution ultrasonography in the diagnosis of scrota1 pathology. I. Normal scrotum and benign disease. J Clin Ultrasound 1993; 21 (6): 355-73. PMID: 8227378.
- Kocakoc E., Serhatlioglu S., Kiris A. et al. Color Doppler sonographic evaluation of inter-relations between diameter, reflux and flow volume of testicular veins in varicocele. Eur J Radiol 2003; 47 (3): 251-6. DOI: https: // doi. org / 10.1016 / S0720-048X (02) 00182-1. PMID: 12927671.
- Капто А.А. Варикозна хвороба органів малого таза у чоловіків. В кн .: Діагностика та лікування веногенной еректильної дисфункції. Під ред. Д.Г. Курбатовa. М .: Медпрактика-М, 2019. С. 140-166. [Kapto AA Varicose disease of the small pelvic organs in men. In: Diagnosis and treatment of venogenic erectile dysfunction. Ed. by DG Kurbatov. M .: Medpraktika-M, 2019. Pp. 140-166. (In Russ.)].
- Felton BM, White JM, Racine MA An uncommon case of abdominal pain: superior mesenteric artery syndrome. West J Emerg Med 2012; 13 (6): 501-2. DOI: 10.5811 / westjem.2012.6.12762. PMID: 23358897.
- Vulliamy P., Hariharan V., Gutmann J., Mukherjee D. Superior mesenteric artery syndrome and the “nutcracker phenomenon”. BMJ Case Rep 2013; 2013. DOI: 10.1136 / bcr-2013-008734. PMID: 23524345. 24.
- Ou-Yang L., Lu GM Underlying anatomy and typing diagnosis of maythurner syndrome and clinical significance an observation based on CT. Spine (Phila Pa 1976) 2016 року; 41 (21): E1284-91. DOI: 10.1097 / BRS.0000000000001765. PMID: 27379417.
- Капто А.А. Ендоваскулярна хірургія клубових вен при двосторонньому варикоцеле і варикозної хвороби вен органів малого таза у чоловіків. Урологічні відомості 2018; 8 (1): 11-7. . DOI: 10.17816 / uroved8111-17.
- Rosen RC, Riley A., Wagner G. et al. The international index of erectile function (IIEF): a multidimensional scale for assessment of erectile dysfunction. Urology 1997; 49 (6): 822-30. PMID: 9187685.
- International Prostate Symptom Score (I-PSS). Available at: https: //www.urospec. com / uro / Forms / ipss.pdf.
- Litwin MS, McNaughton-Collins M., Fowler FJJ et al. The National Institutes of Health chronic prostatitis symptom index: development and validation of a new outcome measure. J Urol 1999; 162 (2): 369-75. PMID: 10411041.
- Coolsaet BL The varicocele syndrome: venography determining the optimal level for surgical management. J Urol 1980; 124 (6): 833-9. DOI: 10.1016 / S0022-5347 (17) 55688-8. PMID: 7441834.
- Bomalaski MD, Mills JL, Argueso LR et al. Iliac vein compression syndrome: an unusual cause of varicocele. J Vasc Surg. 1993; 18 (6): 1064-8. DOI: 10.1016 / 0741-5214 (93) 90564-3. PMID: 8264037.
- Капто А.А. Трансскротальний доступ по лінії Веслінга при оперативному лікуванні варикоцеле (10-річний досвід застосування). Урологічні відомості 2018; 8 (Спец. Вип.): 53-4. .
- Binkert CA, Schoch E., Stuckmann G. et al. Treatment of pelvic venous spur (May-Thurner syndrome) with selfexpanding metallic endoprostheses. Cardiovasc Intervent Radiol 1998; 21 (1): 22-6. DOI: 10.1007 / s002709900205. PMID: 9473541.
- Капто А.А., Виноградов І.В., Харпунов В.Ф., Мамедов Р.Е. Рентгенендоваскулярної ангіопластика і стентування у чоловіка при May- Thurner syndrome. В кн .: Збірник тез 12-го Конгресу Професійної асоціації андрологів України. Сочі, 2019. С. 62..
- Stern JR, Patel VI, Cafasso DE et al. Left-sided varicocele as a rare presentation of May-Thurner syndrome. Ann Vasc Surg 2017; 42: 305.e13-6. DOI: 10.1016 / j.avsg.2016.12.001. PMID: 28258018.
- Marmar JL, DeBenedictis TJ, Praiss D. The management of varicoceles by microdissection of the spermatic cord at the external inguinal ring. Fertil Steril 1985; 43 (4): 583-8. PMID: 3987926.
Стаття опублікована в журналі «Андрологія і генітальна хірургія» випуск №4 2019, стор. 28-38
Журнал
Андрологія і генітальна хірургія 2019 №4
Коментарі
Щоб залишати відгуки необхідно увійти або зареєструватися
Прогноз життя і наслідки захворювання
При своєчасному встановленні діагнозу і адекватному проведенні лікувальних заходів прогноз сприятливий. У хворих вдається досягти прийнятного зростання і розмірів статевих органів. Тривалість життя при відсутності важких анатомічних аномалій з боку інших органів не відрізняється від такої у здорових людей. Жінка з синдромом Тернера-Шерешевського при нормальному розмірі матки може завагітніти і виносити дитину, скориставшись сучасними репродуктивними методами – заплідненням яйцеклітини, взятої від донора, в пробірці (ЕКО).
Сучасні репродуктивні технології – спосіб народити здорову дитину для жінки з генетичним синдромом Тернера-Шерешевського
Особливості статевої Х-хромосоми
Ця Х-хромосома відноситься до однієї з найважливіших в генетичному апараті клітин. Вона найбільша в геномі, в ній міститься до 5% всієї інформації про організм. Її втрата – це летальна мутація, так як разом з нею втрачається до 1700 генів, які регулюють певні життєві процеси. Нормальні клітини жіночого тіла мають дві Х-хромосоми, одна з них неактивна, утворюючи особливу тільце Баррі, з другої активно і постійно зчитується генетична інформація, що реалізується на рівні жіночого тіла. Якщо ж мається на наявність одна Х-хромосома, без другої, дублюючої, можу спостерігатися певні збої з розвитку ще в ембріональному періоді. Вагітність при такій події може нерідко перериватися через загибель зародка, якщо ж дитина росте і розвивається далі, народжується дівчинка з наявністю синдрому Шерешевського.
встановлення діагнозу
У немовляти синдром Шерешевського може виявити неонатолог або педіатр, для цього достатньо провести візуальний огляд. Вказує на наявність патології крилоподібні складки на шиї і набряклі кінцівки. Якщо явні зовнішні симптоми відсутні, то хвороба виявляється в підлітковому віці по низького зросту, відсутності першого менструального кровотечі. Крім того, у хворого слабо виражені вторинні статеві ознаки.
Діагностика обов’язково включає аналіз на гормони. Під час дослідження виявляється збільшення кількості гонадотропінів, зниження рівня естрогенів. Велике діагностичне значення має дослідження і каріотипу. Виявити специфічні ознаки патології лікар може після акушерського УЗД. При підозрі на СШТ призначається пренатальна діагностика:
- УЗД плоду показує, що у нього відсутня носова кістка, збільшена товщина комірцевого простору, недостатньо довгі стегнові або плечові кістки і т. Д. Комплексне дослідження включає біохімію крові матері на β-ХГЛ, РАРР-А.
- Інвазивні дослідження (амніоцентез, біопсія хоріона) є більш точними, ніж вищеописаний скринінг. Пункцію амніотичної оболонки (амніоцентез) і біопсію хоріона (зародкова частина плаценти) найчастіше призначають при підвищеному ризику народження дитини з СШТ, жінкам старше 35 років, а також при поганих результатах неінвазивних досліджень.
Інвазивні дослідження проводять тільки за особливими показниками, вони здатні з високою точністю визначити патологію.
Хворому може знадобитися допомога різних фахівців: генетика, ендокринолога, кардіолога, нефролога, офтальмолога, ЛОР-лікаря, гінеколога, андролога і т. Д.
Щоб виявити вроджені вади і супутні хвороби, призначаються наступні дослідження:
- Ехокардіографія.
- МРТ серця.
- Електрокардіограма.
- УЗД нирок.
- Рентген хребетного стовпа, суглобів кінцівок і решти скелета.
- Денситометрія для перевірки щільності кісток.
- Гінекологічні дослідження.
- УЗД органів малого тазу, мошонки і т. Д.
Щоб відрізнити СШТ від гіпофізарний нанізм (затримка фізичного розвитку внаслідок нестачі соматотропного гормону), потрібно провести аналіз на гормони гіпофіза, рентген турецького сідла (освіта в клиноподібної кістки черепа), ЕЕГ (електроенцефалографію).
Психологічні проблеми дівчаток
Важливим дефектом, типовим для даного синдрому стають своєрідні психічні зміни, пов’язані з дефіцитом статевих гормонів. Вони не формують стану так званої психологічної зрілості. Не варто плутати це зі зниженням інтелекту, це дещо інше поняття, обумовлене впливом гормонів на психіку, зміною поведінки, обумовленого підлогою і подальшими вже дорослими відносинами. Для характеру і поведінки притаманні дитячі звички, немає серйозності і вольових якостей, здатності до самостійного вирішення проблем. Але інтелект при цьому не порушується, здатності до навчання цілком адекватні, є емоційна нестійкість і інфантильність, що створює труднощі у самостійному дорослому житті. Додаються сюди і проблеми закомплексованості, відмінності зовнішності з однолітками істотно тиснуть на психіку, що ускладнює адаптацію в суспільстві і формує комплекси і сором.
Планування вагітності у хворих синдромом Шерешевського – Тернера
Безплідність – дуже важкий симптом, яким страждають практично всі хворі на синдром Тернера – Шерешевського. У більшості випадків пацієнти не можуть мати дітей, проте при мозаїчних формах, коли сформована матка, за допомогою методу екстракорпорального запліднення (ЕКЗ) можливо підсадити або власну запліднену яйцеклітину (за умови повноцінного розвитку яєчників), або донорську.
Використання власних статевих клітин жінки можливо тільки в тому випадку, якщо вони містять в собі нормальний каріотип 46 ХХ.
Екстракорпоральне запліднення – можливість для жінки з генетичним синдромом Шерешевського – Тернера народити здорову дитину
ускладнення
Найчастіше зустрічається такий наслідок захворювання, як жіноче безпліддя. Але відомі випадки, коли жінки, хворі на таку хворобу, можуть завагітніти – традиційно або за допомогою штучного запліднення. Це можливо тільки у хворих з мозаїчним варіантом захворювання або в разі раннього гормонального лікування. В основному ускладнення пов’язані з патологіями внутрішніх органів і систем:
- схильність до раннього розвитку захворювань серцево-судинної системи;
- інфекційно-запальні процеси в нирках і сечовому міхурі;
- отит;
- виникнення злоякісних новоутворень на шкірному покриві;
- різні психологічні проблеми;
- високий ризик розвитку цукрового діабету і ожиріння.
При своєчасній, правильній діагностиці і комплексному лікуванні, люди з таким синдромом повністю адаптуються до життя. Виняток становлять особи з важкими вродженими вадами серця і судин.
Пренатальна діагностика
Ні для кого не секрет, що сучасні методи діагностики дозволяють виявити більшість генетичних відхилень плоду задовго до народження. Одну з таких процедур, неінвазивний пренатальне тестування (скринінг позаклітинної ДНК плоду, що знаходиться в крові матері), можна виробляти з кінця першого триместру вагітності.
Перевагою цієї процедури є те, що вона абсолютно безпечна для матері і плоду. Крім синдрому Тернера, скринінг позаклітинної ДНК дозволяє виявити такі анеуплоїдії як синдром Патау (трисомія по 13-й хромосомі), синдром Едвардса (трисомія по 18-й хромосомі), синдром Дауна (трисомія по 21-й хромосомі), трисомія по Х-хромосомі , синдром Клайнфельтера, синдром Мартіна-Белл.
За допомогою ультразвукового дослідження можна виявити аномалії розвитку нирок і серця – вони відносяться до можливих симптомів захворювання.
Синдром можна виявити за допомогою амніоцентезу або біопсії хоріона. Обидві ці процедури інвазивних і мають протипоказання. До їх достоїнств відноситься висока точність отриманого результату.
методи лікування
гормональна терапія
Основними завданнями лікування синдрому Тернера-Шерешевського є досягнення прийнятного зростання і адекватне протягом статевого дозрівання. Перша вирішується за допомогою призначення гормону росту – Соматотропіну аж до остаточного закриття хрящових зон довгих кісток верхніх і нижніх кінцівок. З 12-річного віку для запуску в організмі статевого дозрівання, зростання молочних залоз і матки, становлення менструального циклу призначаються жіночі статеві гормони – естрогени, потім і Прогестерон. Препарати приймаються жінкою з синдромом Тернера-Шерешевського в середньому до 50 років.
Людський гормон росту – основний метод лікування синдрому Тернера-Шерешевського
хірургічне лікування
Оперативне лікування проводиться в наступних випадках:
- супутній вроджений порок серця;
- необхідність корекції деформації хребта;
- корекція крилоподібних складок на шиї з косметичною метою за допомогою методів пластичної хірургії;
немедикаментозне лікування
До немедикаментозного лікування синдрому Тернера-Шерешевського відносяться такі заходи:
- раціональний режим праці і відпочинку;
- дієта із зменшеною кількістю вуглеводів, збагачена овочами, фруктами і вітамінами;
- лікувальний масаж;
- лікувальна гімнастика;
- електрофорез і магнітотерапія;
- санаторно-курортне лікування;
Народні кошти не довели свою ефективність в боротьбі з даним захворюванням.
причини
Причини виникнення моносомии по Х-хромосомі пов’язані з порушенням поділу клітин і формування генетичного матеріалу в період внутрішньоутробного розвитку. Патологічні порушення виникають в ранній період розвитку плода.
Через нестачу або неправильного розподілу молекул ДНК порушується структура окремих хромосом. При СШТ пошкоджується Х-хромосома. При класичній формі хвороби її немає – 45 ХО (норма – 46ХХ). Через дефіцит генетичного матеріалу розвиваються специфічні симптоми, які знижують якість життя дитини відразу після появи на світ.
Важливо. Синдром Шерешевського-Тернера, на відміну від інших хромосомних патологій, не загрожує життю. Деякі жінки навіть можуть завагітніти і народити дитину.
Медики виділяють наступні передумови для виникнення СШТ:
- Інфекційні захворювання перед вагітністю або під час неї.
- Вплив на майбутню матір радіації або електромагнітних полів.
- Вплив хімічних речовин на плід (ранні стадії розвитку).
- Генетична схильність.
- Виснаження, недостатнє харчування під час вагітності.
Синдром Шерешевського може розвиватися навіть при відсутності вищеописаних факторів.
Патологія може розвиватися за різними сценаріями: друга хромосома відсутня повністю, спостерігається часткова моносомія, тобто Х-хромосома відсутня в повному обсязі, в ній відбуваються різноманітні перебудови. При СШТ можливий мозаицизм, коли в тканинах присутні генетично розрізняються клітини.
Моносомія по 45, Х хромосомі – це найпоширеніший каріотип. Найчастіше неушкоджена хромосома материнська. Цей різновид моносомии виникає через те, що хроматиди не розходяться внаслідок поділу клітин в батьківській гамете.
При мозаїчній формі моносаміі (45, Х0 / 46, ХХ; 45, Х0 / 46, ХУ і т. Д.) Частина клітин містять одну Х-хромосому, а інші – дві, при цьому друга може бути Х або У. Цей різновид патології має легший перебіг.
Хвороба може виникати через те, що одна Х-хромосома інактивована. Як правило, це відбувається в період раннього внутрішньоутробного розвитку. Цей процес спонтанний і діє на будь-які пари хромосом, але зазвичай страждає саме х-хромосома. Деякі гени уникають інактивації.
У чоловіків СШТ виникає внаслідок перенесення ділянки хромосоми на негомологічну хромосому або в результаті мозаїцизму.
Ризик розвитку патології не залежить від віку вагітної. Кількісні, якісні і структурні патології Х-хромосоми виникають через те, що порушується мейотическое розподіл хромосом (Х-моносомія) або дроблення зиготи (мозаїцизм). У більшості випадків при каріотипі 45, Х0 інактивується батьківська хромосома.
Через відсутність Х-хромосоми або її структурних дефектів порушується формування статевих залоз, а також різноманітні аномалії розвитку.
Практично завжди майбутня мати, яка виношує плід, який має СШТ, страждає від токсикозу, загрози мимовільного аборту або передчасних пологів.
профілактичні заходи
Хромосомна патологія діагностується у дітей здорових батьків. Передбачити можливість розвитку синдрому неможливо, як і вдатися до якихось заходів профілактики.
Єдиний спосіб виявити нестачу Х-хромосоми – пройти пренатальну діагностику на початкових термінах вагітності. На 11-13 тижні жінка може здати кров на наявність в ній плазмового протеїну А. Це означає підвищену ймовірність генетичної аномалії. Підтвердити або спростувати діагноз можна за допомогою біопсії ворсинок хоріона. За ним визначають каріотип плода, структуру хромосом.
Якщо синдром виявляється на стадії виношування, не потрібно панікувати. Дівчинка може народитися без зовнішніх проявів захворювання, а недорозвиненість статевих органів коригується за допомогою гормональної терапії.
Loading …
Поділіться з друьямі!
Наявність функціональних розладів при синдромі
Крім зовнішніх дефектів у вигляді дітей з цим синдромом, є також і особливості поведінки і роботи внутрішніх органів, які обумовлені наявністю тільки однієї Х-хромосоми. Їх відзначають самі батьки у малюків, починаючи з найперших тижнів їх життя при догляді. Якщо говорити про найбільш частих функціональних дефектах, до них можна віднести:
- Розлад рефлексу смоктання, пов’язане з наявністю як аномалій в будові зубочелюстного апарату, так і при проблемах з нервовою системою і тонусом м’язів обличчя. Навіть при відсутності аномалій у розвитку порожнини рота і неба, можуть виникати неврологічні розлади з проблемами контролю за тонусом м’язів обличчя.
- Напади загального занепокоєння, при якому немає характерних і типових проявів, дитина просто перебуває в збудженому і неспокійному стані тривалий час. В цілому, крихітка погано спить і не завжди реагує на голоси батьків, звернені до неї, може багато і безпричинно плакати. У міру розвитку навичок і дозрівання нервової системи ці прояви поступово проходять з віком, коли формуються нові рефлекси і навички.
- Часті і активні відрижки їжі, що виникають після годування. Нерідко відрижки виникають через кілька хвилин або годин після годування, і нерідко подібні проблеми можуть вказувати на аномалії в будові травної трубки (різкому звуженні стравоходу, кишечника). Але типові відрижки і без будь-яких проблем в будові травної системи, а тільки за рахунок нерівномірного скорочення м’язів в кишкової трубці, через що страждає перистальтика. Це загрожує розладом проходження харчової грудки вниз, до товстого кишечнику, що призводить до затримки їжі в шлунку і її відрижки. За мерее дорослішання відрижки проходять, вони змінюються наполегливими і тривалими запорами або їх чергуванням з проносами.
- Запізніле формування мовних навичок пов’язано нерідко з затримкою розумового розвитку або проблемою тонусу м’язів в порожнині рота, що ускладнює освоєння мови. Типово подібне для 20% дітей.
- Нерідко у дівчаток спостерігається тривалий нетримання сечі, пов’язане з порушенням іннервації сечового міхура. Нерідко проблема енурезу не проходить до шкільного віку і навіть підліткового, потім вона зникає.
Необхідні діагностичні дослідження
Виявити захворювання представляється можливим ще до народження дитини, завдяки сучасним методам генетичного аналізу. Тому всі обстеження прийнято ділити на пренатальні і постнатальні. До діагностиці, використовуваної до народження дитини, відносять:
- Каріотипування майбутніх батьків, тобто аналіз їх ДНК. Наявність дефектів в матеріалі матері або батька збільшує ризик розвитку патології.
- Хоча за допомогою УЗД і неможливо поставити точний діагноз, воно широко застосовується для виявлення непрямих проявів синдрому. Набряклість плода, аномалії розвитку скелета, пороки серця і інші ознаки вказують на формування генетичного захворювання.
- Найточнішим є проведення каріотипування малюка. Для цього застосовуються різні інвазивні методики, що дозволяють отримати генетичний матеріал дитини. Дані процедури пов’язані з ризиком інфікування та переривання вагітності, тому рішення про їх проведення приймається спільно з гінекологом і генетиком.
Після появи дитини на світ поставити діагноз, як правило, не складає труднощів. Це пов’язано з тим, що в результаті розвитку синдрому Шерешевського-Тернера у пацієнта формуються специфічні симптоми, які вказують на проблему. Для підтвердження наявності захворювання проводяться такі тести:
- Аналізи крові і сечі використовуються для визначення гормонального статусу малюка, а також оцінки роботи внутрішніх органів.
- Для виключення наявності вроджених кардіальних уражень здійснюється УЗД, в ході якого робляться своєрідні фото серця і його виміри, а також ЕКГ.
різновиди патології
- відсутність Х-хромосоми.
Ця форма є найбільш важкою і часто зустрічається варіантом (близько 60% випадків захворювання). Вона характеризується повною відсутністю статевої хромосоми, що призводить до вираженої класичної клінічної картині. Генетичного матеріалу не достатньо для повноцінного розвитку плоду і починаючи з 3 місяців вагітності, спочатку нормальні яєчники зазнають змін. Їх структура заміщується сполучною тканиною, у плода з’являються вади розвитку;
- мозаїчна форма.
На цей варіант хвороби припадає близько 20% від загального числа захворювання. Недуга протікає більш сприятливо, оскільки частина клітин має нормальний набір хромосом, а значить компенсує прояви патології. Зовнішність дівчаток з мозаїчною формою захворювання може відповідати класичних проявів синдрому, хоча типові ознаки менш виражені. Грубі порушення в структурі і функції внутрішніх органів виявляються рідше, що значно полегшує перебіг недуги;
- порушення будови Х-хромосоми.
20% хворих спадковим синдромом мають спочатку нормальний набір хромосом, але одна з них відрізняється серйозними порушеннями структури молекули. У таких випадках у жінок спостерігаються ознаки, характерні для класичної форми захворювання, але вираженість їх значно менше.
У літературі описані поодинокі випадки виявлення синдрому Шерешевського-Тернера у чоловіків. Це пояснюється хромосомним мозаїцизмом, коли в організмі присутні клітини з різним набором молекул ДНК.
пов’язані порушення
Симптоми наступних розладів можуть бути подібними з симптомами синдрому Тернера. Порівняння корисні для диференціальної діагностики.
Синдром Нунана – генетичне захворювання, яке проявляється при народженні (вроджений). Розлад характеризується широким спектром симптомів і фізичних особливостей, які сильно розрізняються за діапазоном і ступеня тяжкості.
У багатьох порушених осіб асоційовані аномалії включають:
- характерний зовнішній вигляд особи;
- широку або перетинчасту шию;
- низьку лінію волосся;
- типову деформацію грудної клітки;
- низький ріст.
Характерні аномалії головного та лицьової (черепно-лицевої) області включають:
- широко розставлені очі (очної гипертелоризм);
- складки шкіри, що покривають внутрішні кути очей (епікантальние складки);
- опущені верхні повіки (птоз);
- маленьку щелепу (микрогнатия);
- продавлений корінь носа;
- короткий ніс з широкою основою;
- низькорозташованим вуха (pinnae).
Присутні відмінні скелетні мальформації, такі як аномалії грудини, викривлення хребта (кіфоз, сколіоз), зовнішнє відхилення ліктів (cubitus valgus). У багатьох немовлят з синдромом Ноонана є дефекти серця, такі як обструкція кровотоку з нижньої правої камери в легені (легеневий клапанний стеноз).
Додаткові аномалії включають вади розвитку судин, лімфи, погане згортання крові, дефіцит тромбоцитів, труднощі навчання, помірну інтелектуальну інвалідність, крипторхізм на першому році життя у уражених чоловіків і інші симптоми.
Дізнатися більше Ознаки діагностика і лікування синдрому Ротора
Синдром Нунан є аутосомно-домінантним генетичним розладом, викликаним аномаліями (мутаціями) чотирьох основних генів. Деякі симптоми можуть зовні нагадувати симптоми синдрому Тернера (низький зріст, перетинкова шия). Однак між цими двома розладами існує багато важливих відмінностей.
Синдром Нунана вражає як чоловіків, так і жінок, існує нормальний хромосомний каріотип. Тільки жінки схильні до впливу синдрому Тернера, який характеризується порушеннями, що впливають на Х-хромосому.
Медичний курйоз
Існують ще два захворювання, які іноді скорочено називають синдромами Тернера. Відкрили і описали ці патології однофамільці американського ендокринолога Генрі Тернера, звідси і плутанина з назвами. Це синдроми Мея-Тернера і Персонейджа-Тернера. Обидва ці захворювання ніяк не пов’язані з моносомією по Х-хромосомі, описаної вище, і навіть не є спадковими хворобами.
Перший опис синдрому Мея-Тернера з’явилося в 1957 році. Патологія проявляється порушенням відтоку венозної крові від лівої нижньої кінцівки і органів таза, в результаті чого хворі страждають від постійних болів в лівій нозі і області тазу. На пізніх стадіях хвороби на венографии видно тромбоз глибоких вен. Синдром складно діагностується, особливо на ранніх стадіях, так як спочатку протікає безсимптомно. Синдром Мея-Тернера зазвичай починається в підлітковому віці, у чоловіків зустрічається частіше, ніж у жінок.

В якості основної діагностичної процедури застосовується УЗД клубових вен таза. У разі наявності захворювання, ліва загальна клубова вена в діаметрі значно більше нормальної. Для підтвердження діагнозу застосовується магнітно-резонансна ангіографія клубових вен з контрастом. На пізніх стадіях хвороби лікування проводиться хірургічно. Часто призначається прийом медичних препаратів, покликаних відновити нормальний кровотік.
Синдром Персонейджа-Тернера в клінічній практиці зустрічається рідко і його причини досі точно не встановлені. Перший сигнал про наявність захворювання – різка безпричинна біль в плечі або руці, рідше – в обох руках одночасно. Багато людей, у яких вперше проявилася ця патологія, не вважають за потрібне звертатися за допомогою до фахівця, сподіваючись, що симптоми зникнуть самі.

Біль може не проходити кілька днів, іноді кілька тижнів. Біль посилюється при рухах і слабшає, якщо кінцівка знаходиться в спокої. Багато пацієнтів перестають розробляти руку, через що з часом розвивається м’язова дистрофія. У більшості людей захворювання з часом проходить без будь-якого втручання. Іноді потрібно прийом сильних анальгетиків. Причини синдрому Персонейджа-Тернера представляють великий інтерес для медиків, але поки не вивчені. Поширеність захворювання також не відома.